Visium HD 3’ (peak)#
This notebook demonstrates essential steps for analyzing spatial transcriptome 3’ end diversity (TREND) using Visium HD 3’ sequencing data (peak-level).
Load preprocessed peak-level quantification.
Run FFT-accelerated spatial variability tests with
SplisosmFFT.Compare SV test results across spatial resolutions.
Compare results across spatial resolutions and with
SplisosmNP.
For differential usage testing with SplisosmFFT, see the Visium HD FFPE (probe) Part II tutorial.
Estimated runtime: ~15 min.
Preliminary notes#
3’ end sequencing data captures variation near the 3’ end of transcripts (alternative last exon, alternative polyadenylation etc.) as well as around intronic poly-A-rich regions. We jointly refer to these as transcriptome 3’ end diversity (TREND) events.
To run the analysis, we first call junction-aware peaks from the BAM file using Sierra, then quantify peak-level expression in each Visium HD bin using UMI-tools. An example workflow is provided in scripts/visiumhd_3p_trend_quant.sh under the scripts directory, which generates a peak-level SpatialData object stored as a .zarr file.
Imports#
from __future__ import annotations
from itertools import combinations
from pathlib import Path
import warnings
import annsel as an
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
from scipy.stats import spearmanr
import spatialdata as sd
import spatialdata_plot
from spatialdata import rasterize_bins
from splisosm import SplisosmFFT, SplisosmNP
from splisosm.utils import counts_to_ratios
warnings.filterwarnings("ignore", category=FutureWarning)
plt.rcParams["figure.dpi"] = 120
plt.rcParams["figure.figsize"] = (6, 4)
Configure paths and core parameters#
# Path to the preprocessed peak-level SpatialData zarr
sdata_zarr = Path("path/to/sdata_peak.filtered.zarr")
# Optional path to Sierra-called peaks in BED12 format (for visualization)
bed12_file = Path("path/to/peak.txt.bed12")
# Optional path to GTF annotation (for visualization)
gtf_file = Path("path/to/gencode.vM33.annotation.gtf.gz")
# Dataset and table identifiers (16 µm binning used throughout)
dataset_id = ""
test_table = "square_016um"
test_bins_element = f"{dataset_id}_square_016um"
# Peak annotation column names
group_iso_by = "gene_ids"
gene_name_col = "gene_ids"
candidate_gene_name_cols = ["gene_name", "gene_ids", "gene_symbol", "gene_names"]
# Peak QC filters
min_counts = 10
min_bin_pct = 0.01
Load preprocessed peak-level SpatialData#
In this example, we use a Visium HD 3’ mouse brain dataset (fresh frozen) provided by 10x Genomics. We load the preprocessed, peak-level SpatialData object generated by our custom quantification workflow (scripts/visiumhd_3p_trend_quant.sh). The resulting Sierra peak quantification matrix is located at outs/binned_outputs/square_002um/raw_probe_bc_matrix.h5.
%%time
if sdata_zarr.exists():
print("Loading preprocessed SpatialData...")
sdata = sd.read_zarr(sdata_zarr)
else:
print("Building peak-level SpatialData from quantification outputs...")
from splisosm.io import load_visiumhd_probe
sdata = load_visiumhd_probe(
path='path/to/outs',
bin_sizes=bin_sizes,
filtered_counts_file=True,
load_all_images=False,
var_names_make_unique=True,
counts_layer_name="counts",
)
# Optional: cache for future runs
# sdata.write(sdata_zarr)
sdata
Loading preprocessed SpatialData...
CPU times: user 6.62 s, sys: 1.31 s, total: 7.93 s
Wall time: 5.1 s
SpatialData object, with associated Zarr store: /Users/jysumac/Projects/SPLISOSM_paper/data/visiumhd_3p_mouse_cbs/sdata_peak.filtered.zarr
├── Images
│ ├── '_hires_image': DataArray[cyx] (3, 5492, 6000)
│ └── '_lowres_image': DataArray[cyx] (3, 549, 600)
├── Shapes
│ ├── '_cell_segmentations': GeoDataFrame shape: (84031, 2) (2D shapes)
│ ├── '_square_002um': GeoDataFrame shape: (5998466, 1) (2D shapes)
│ ├── '_square_008um': GeoDataFrame shape: (376419, 1) (2D shapes)
│ └── '_square_016um': GeoDataFrame shape: (94592, 1) (2D shapes)
└── Tables
├── 'cell_segmentations': AnnData (84031, 19575)
├── 'square_002um': AnnData (5998466, 19575)
├── 'square_008um': AnnData (376419, 19575)
└── 'square_016um': AnnData (94592, 19575)
with coordinate systems:
▸ '', with elements:
_hires_image (Images), _lowres_image (Images), _cell_segmentations (Shapes), _square_002um (Shapes), _square_008um (Shapes), _square_016um (Shapes)
▸ '_downscaled_hires', with elements:
_hires_image (Images), _cell_segmentations (Shapes), _square_002um (Shapes), _square_008um (Shapes), _square_016um (Shapes)
▸ '_downscaled_lowres', with elements:
_lowres_image (Images), _cell_segmentations (Shapes), _square_002um (Shapes), _square_008um (Shapes), _square_016um (Shapes)
SPLISOSM can be run at any spatial resolution, but data become sparser at finer scales.
def summarize_table(adata):
X = adata.layers["counts"] if "counts" in adata.layers else adata.X
if hasattr(X, "nnz"):
nnz = int(X.nnz)
total = int(X.shape[0] * X.shape[1])
density = nnz / total if total else np.nan
else:
arr = np.asarray(X)
nnz = int(np.count_nonzero(arr))
total = int(arr.size)
density = nnz / total if total else np.nan
return {
"n_peaks": int(adata.n_vars),
"n_bins": int(adata.n_obs),
"count_mtx_density": density,
}
rows = []
for key in sorted(sdata.tables.keys()):
if key.startswith("square_"):
rows.append({"table": key, **summarize_table(sdata.tables[key])})
table_summary = pd.DataFrame(rows).sort_values("table")
table_summary
| table | n_peaks | n_bins | count_mtx_density | |
|---|---|---|---|---|
| 0 | square_002um | 19575 | 5998466 | 0.000437 |
| 1 | square_008um | 19575 | 376419 | 0.006380 |
| 2 | square_016um | 19575 | 94592 | 0.022677 |
To balance sparsity and computation, we recommend 16µm or 8µm bins for initial analysis. Resolution comparison is shown at the end of this notebook.
# Optional: inspect available coordinate systems and image/shape keys
print("Tables:", sorted(sdata.tables.keys()))
print("Images:", sorted(getattr(sdata, "images", {}).keys()))
print("Shapes:", sorted(getattr(sdata, "shapes", {}).keys()))
# Quick guardrails before model setup
if test_table not in sdata.tables:
raise ValueError(f"{test_table} is not available. Choose from: {sorted(sdata.tables.keys())}")
adata_test = sdata.tables[test_table]
if group_iso_by not in adata_test.var.columns:
raise ValueError(f"{group_iso_by} not found in {test_table}.var columns")
if gene_name_col not in adata_test.var.columns:
for col in candidate_gene_name_cols:
if col in adata_test.var.columns:
gene_name_col = col
break
print(f"Using table={test_table}, bins={test_bins_element}")
print(f"Grouping column={group_iso_by}, display names={gene_name_col}")
Tables: ['cell_segmentations', 'square_002um', 'square_008um', 'square_016um']
Images: ['_hires_image', '_lowres_image']
Shapes: ['_cell_segmentations', '_square_002um', '_square_008um', '_square_016um']
Using table=square_016um, bins=_square_016um
Grouping column=gene_ids, display names=gene_ids
The features in this dataset represent peaks (spanning single exons or splice junctions) identified using Sierra’s FindPeaks function. These peaks were subsequently annotated based on their overlap with coding sequences (CDS), untranslated regions (UTR3 and UTR5), or intronic regions using AnnotatePeaksFromGTF(annotation_correction=FALSE).
sdata.tables[test_table].var.head(5)
| gene_ids | probe_ids | feature_types | CDS | Junctions | UTR3 | UTR5 | end | exon | genome | intron | seqnames | start | strand | width | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0610009B22Rik:chr11:51576213-51576468:-1 | 0610009B22Rik | 0610009B22Rik:chr11:51576213-51576468:-1 | Gene Expression | no-junctions | YES | 51576468 | YES | chr11 | 51576213 | - | 256 | ||||
| 0610009L18Rik:chr11:120241627-120242029:1 | 0610009L18Rik | 0610009L18Rik:chr11:120241627-120242029:1 | Gene Expression | no-junctions | 120242029 | YES | chr11 | 120241627 | + | 403 | |||||
| 0610030E20Rik:chr6:72329741-72330131:1 | 0610030E20Rik | 0610030E20Rik:chr6:72329741-72330131:1 | Gene Expression | no-junctions | YES | 72330131 | YES | chr6 | 72329741 | + | 391 | ||||
| 0610040B10Rik:chr5:143318096-143318420:1 | 0610040B10Rik | 0610040B10Rik:chr5:143318096-143318420:1 | Gene Expression | no-junctions | 143318420 | YES | chr5 | 143318096 | + | 325 | |||||
| 0610040J01Rik:chr5:64056587-64056953:1 | 0610040J01Rik | 0610040J01Rik:chr5:64056587-64056953:1 | Gene Expression | no-junctions | YES | 64056953 | YES | chr5 | 64056587 | + | 367 |
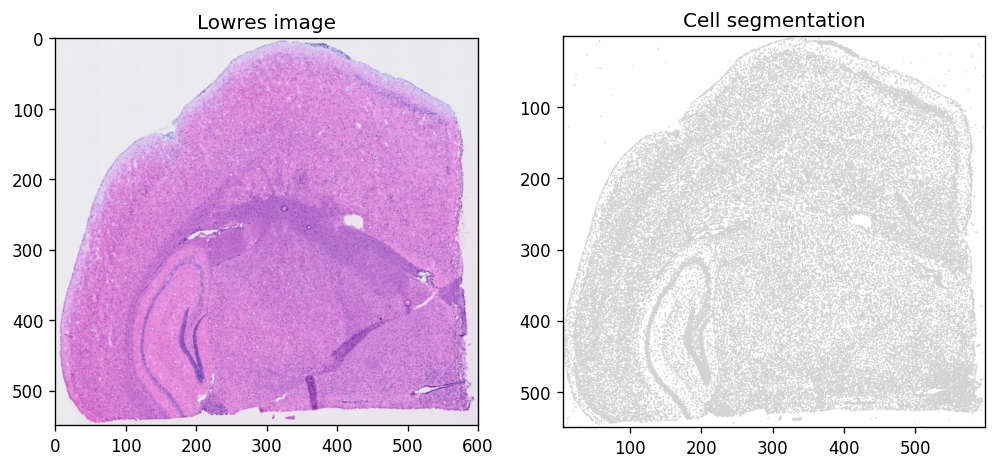
Use spatialdata-plot to visualize tissue morphology:
%%time
axes = plt.subplots(1, 2, figsize=(10, 5))[1].flatten()
sdata.pl.render_images(f"{dataset_id}_lowres_image").pl.show(
coordinate_systems=f"{dataset_id}_downscaled_lowres",
ax=axes[0], title="Lowres image"
)
sdata.pl.render_shapes(f"{dataset_id}_cell_segmentations").pl.show(
coordinate_systems=f"{dataset_id}_downscaled_lowres",
ax=axes[1], title="Cell segmentation"
)
CPU times: user 3.23 s, sys: 145 ms, total: 3.38 s
Wall time: 3.42 s
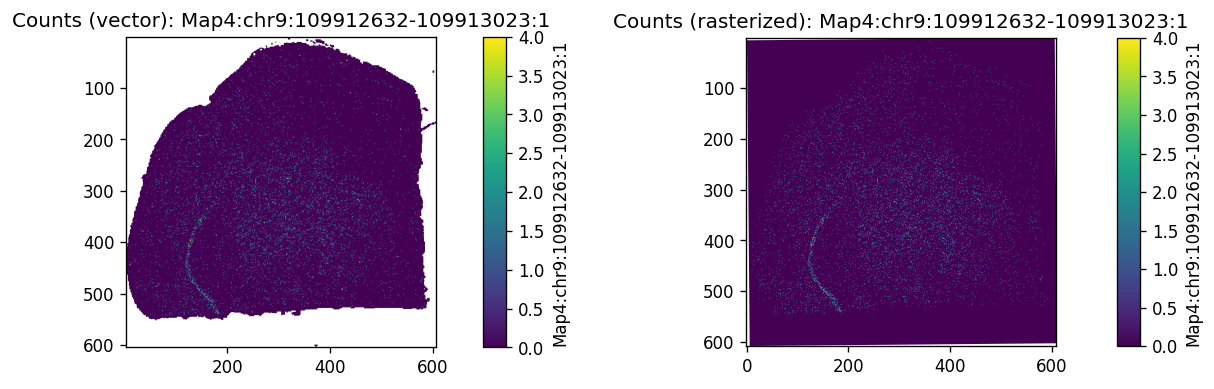
To visualize peak expression as an image, rasterize the spatial bins first:
%%time
for bin_size in ["016", "008", "002"]:
# rasterize_bins() requires a compressed sparse column (csc) matrix
sdata.tables[f"square_{bin_size}um"].X = sdata.tables[f"square_{bin_size}um"].X.tocsc()
rasterized = rasterize_bins(
sdata,
f"{dataset_id}_square_{bin_size}um",
f"square_{bin_size}um",
"array_col",
"array_row",
)
sdata[f"rasterized_{bin_size}um"] = rasterized
CPU times: user 3.65 s, sys: 622 ms, total: 4.27 s
Wall time: 4.34 s
Then visualize a peak’s global expression at 16µm resolution:
%%time
peak_name = 'Map4:chr9:109912632-109913023:1'
print(f"Using example feature: {peak_name}")
axes = plt.subplots(1, 2, figsize=(10, 5))[1].flatten()
sdata.pl.render_shapes(f"{dataset_id}_square_016um", color=peak_name).pl.show(
coordinate_systems=f"{dataset_id}_downscaled_lowres",
ax=axes[0], title=f"Counts (vector): {peak_name}"
)
sdata.pl.render_images(f"rasterized_016um", channel=peak_name).pl.show(
coordinate_systems=f"{dataset_id}_downscaled_lowres",
ax=axes[1], title=f"Counts (rasterized): {peak_name}"
)
plt.subplots_adjust(wspace=1)
plt.show()
Using example feature: Map4:chr9:109912632-109913023:1
CPU times: user 5.46 s, sys: 1.27 s, total: 6.74 s
Wall time: 7 s
Spatial variability testing with SplisosmFFT#
To detect peak usage variation within a given gene, we run spatial variability test using hsic-ir.
Filtering and model setup#
Peak filtering criteria:
min_counts: minimum total UMI count across all bins.min_bin_pct: minimum fraction of bins in which a peak must be detected.Genes with fewer than two passing peaks are automatically excluded.
model = SplisosmFFT(neighbor_degree=1, rho=0.99)
model.setup_data(
sdata=sdata,
bins=test_bins_element,
table_name=test_table,
col_key="array_col",
row_key="array_row",
layer="counts",
group_iso_by=group_iso_by, # 'gene_ids'
gene_names=gene_name_col, # 'gene_ids'
min_counts=min_counts,
min_bin_pct=min_bin_pct,
filter_single_iso_genes=True
)
model
=== SplisosmFFT
- Number of genes: 1061
- Number of spots: 94592
- Number of spots after rasterization: 134688
- Number of covariates: 0
- Average isoforms per gene: 2.2
=== Model configurations
- Neighborhood degree: 1
- Spatial autocorrelation rho: 0.99
=== Test results
- Spatial variability: N/A
- Differential usage: N/A
Extract gene-level summary statistics:
%%time
gene_meta = model.extract_feature_summary(level='gene')
gene_meta.sort_values('perplexity', ascending=False).head(5)
Genes: 100%|██████████| 1061/1061 [00:00<00:00, 1509.27it/s]
CPU times: user 771 ms, sys: 119 ms, total: 889 ms
Wall time: 890 ms
| n_isos | perplexity | pct_bin_on | count_avg | count_std | major_ratio_avg | |
|---|---|---|---|---|---|---|
| gene | ||||||
| Ppp3ca | 6 | 4.951889 | 0.407138 | 0.869429 | 1.597794 | 0.307328 |
| Celf4 | 5 | 3.819581 | 0.202174 | 0.250539 | 0.552908 | 0.477362 |
| Homer1 | 4 | 3.788384 | 0.063769 | 0.070535 | 0.283515 | 0.364508 |
| Pcmt1 | 4 | 3.785652 | 0.139145 | 0.161441 | 0.432617 | 0.378561 |
| Rbbp6 | 4 | 3.780505 | 0.081297 | 0.087640 | 0.305624 | 0.332931 |
Peak-level summary is also available:
peak_meta = model.extract_feature_summary(level='isoform')
peak_meta.sort_values('gene_ids', ascending=False).head(5)
| gene_ids | probe_ids | feature_types | CDS | Junctions | UTR3 | UTR5 | end | exon | genome | ... | start | strand | width | pct_bin_on | count_total | count_avg | count_std | ratio_total | ratio_avg | ratio_std | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| mt-Nd5:chrM:12597-13503:1 | mt-Nd5 | mt-Nd5:chrM:12597-13503:1 | Gene Expression | YES | no-junctions | 13503 | YES | ... | 12597 | + | 907 | 0.295427 | 35035.0 | 0.370380 | 0.642801 | 0.672909 | 0.670371 | 0.432255 | |||
| mt-Nd5:chrM:11870-13196:1 | mt-Nd5 | mt-Nd5:chrM:11870-13196:1 | Gene Expression | YES | no-junctions | 13196 | YES | ... | 11870 | + | 1327 | 0.135720 | 14026.0 | 0.148279 | 0.391148 | 0.269394 | 0.271458 | 0.408964 | |||
| mt-Nd5:chrM:11742-12338:1 | mt-Nd5 | mt-Nd5:chrM:11742-12338:1 | Gene Expression | YES | no-junctions | 12338 | YES | ... | 11742 | + | 597 | 0.031123 | 3004.0 | 0.031757 | 0.178934 | 0.057697 | 0.058170 | 0.215502 | |||
| mt-Nd4:chrM:11161-11413:1 | mt-Nd4 | mt-Nd4:chrM:11161-11413:1 | Gene Expression | YES | no-junctions | 11413 | YES | ... | 11161 | + | 253 | 0.906726 | 332118.0 | 3.511058 | 2.643438 | 0.937358 | 0.936997 | 0.150768 | |||
| mt-Nd4:chrM:11100-11519:1 | mt-Nd4 | mt-Nd4:chrM:11100-11519:1 | Gene Expression | YES | across-junctions | 11519 | YES | ... | 11100 | + | 420 | 0.168820 | 17843.0 | 0.188631 | 0.443044 | 0.050359 | 0.050763 | 0.136139 |
5 rows × 22 columns
Running HSIC-IR test#
%%time
model.test_spatial_variability(
method="hsic-ir",
ratio_transformation="none",
n_jobs=-1,
print_progress=True,
)
sv_res_16um = model.get_formatted_test_results(
"sv", with_gene_summary=True
).sort_values("pvalue_adj")
SV [hsic-ir]: 100%|██████████| 76/76 [00:02<00:00, 33.65it/s]
CPU times: user 9.39 s, sys: 2.86 s, total: 12.2 s
Wall time: 2.92 s
Top genes, sorted by adjusted p-value:
sig_001 = int((sv_res_16um["pvalue_adj"] < 0.01).sum())
print(
"Spatially variably processed genes (FDR < 0.01, 16 µm): "
f"{sig_001} out of {sv_res_16um.shape[0]} total genes"
)
sv_res_16um.head(5)
Spatially variably processed genes (FDR < 0.01, 16 µm): 506 out of 1061 total genes
| gene | statistic | pvalue | pvalue_adj | n_isos | perplexity | pct_bin_on | count_avg | count_std | major_ratio_avg | |
|---|---|---|---|---|---|---|---|---|---|---|
| 811 | Septin8 | 1.398869e-06 | 0.0 | 0.0 | 3 | 2.184668 | 0.113213 | 0.125941 | 0.372038 | 0.707295 |
| 382 | Gpm6b | 1.697766e-06 | 0.0 | 0.0 | 2 | 1.544330 | 0.348698 | 0.466562 | 0.747658 | 0.843043 |
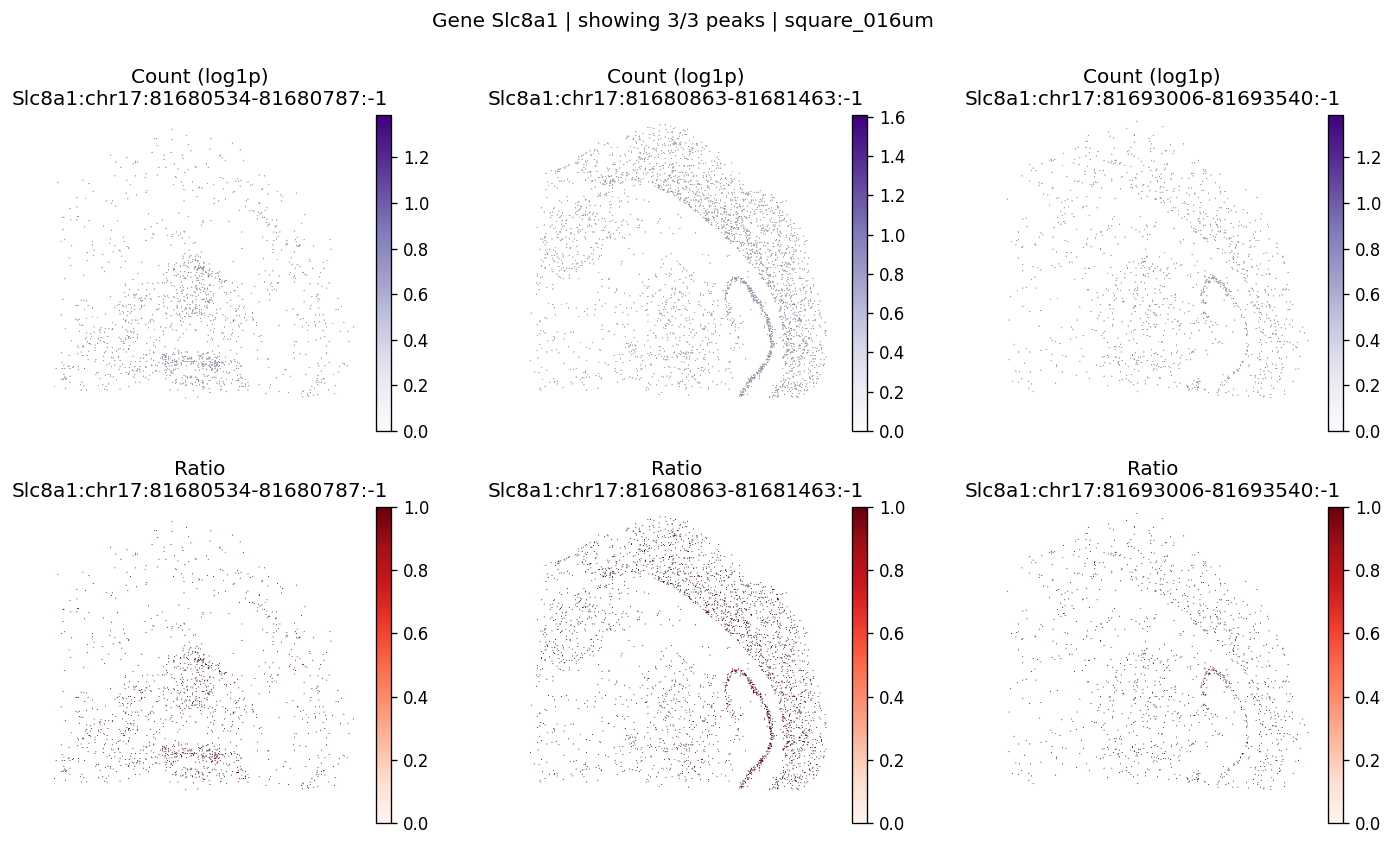
| 840 | Slc8a1 | 6.530548e-07 | 0.0 | 0.0 | 3 | 2.737542 | 0.059466 | 0.065788 | 0.274655 | 0.541539 |
| 357 | Gls | 6.126092e-07 | 0.0 | 0.0 | 2 | 1.514386 | 0.124408 | 0.143976 | 0.411355 | 0.854395 |
| 138 | Camk1d | 7.656467e-07 | 0.0 | 0.0 | 2 | 1.905026 | 0.091213 | 0.101795 | 0.340931 | 0.654689 |
Visualize significant events#
The helper below renders per-peak log1p counts and within-gene ratios on the testing grid.
def ensure_rasterized(sdata, bin_table: str, bin_element: str, layer: str = "counts"):
raster_key = f"rasterized_{bin_table}_{layer}"
if raster_key in sdata.images:
return raster_key
adata = sdata.tables[bin_table]
adata.X = adata.layers[layer]
if hasattr(adata.X, "tocsc") and getattr(adata.X, "format", None) != "csc":
adata.X = adata.X.tocsc()
sdata[raster_key] = rasterize_bins(
sdata,
bins=bin_element,
table_name=bin_table,
col_key="array_col",
row_key="array_row",
)
return raster_key
The plotting function checks peak availability, lazily rasterizes data on demand, computes log1p counts and within-gene ratios, and optionally masks zero values.
def plot_gene_peak_maps(
sdata,
bin_table: str,
bin_element: str,
gene_id: str,
peak_meta: pd.DataFrame | None = None,
group_col: str = "gene_ids",
max_peaks: int = 4,
hide_zero_count: bool = True,
hide_zero_ratio: bool = True,
):
adata = sdata.tables[bin_table]
if peak_meta is None:
peak_meta = adata.var.copy()
if group_col not in peak_meta.columns:
raise ValueError(f"'{group_col}' not found in peak_meta columns")
peak_names = peak_meta.index[
peak_meta[group_col].astype(str) == str(gene_id)
].tolist()
if len(peak_names) == 0:
raise ValueError(f"No peaks found for gene id '{gene_id}'")
if any(peak not in adata.var_names for peak in peak_names):
raise ValueError(f"Some peaks not found in {bin_table}.var_names")
n_peaks = len(peak_names)
n_shown = min(n_peaks, max_peaks)
peak_names = peak_names[:n_shown]
raster_key = ensure_rasterized(sdata, bin_table=bin_table, bin_element=bin_element)
data = sdata[raster_key].sel(c=peak_names).values
counts_cube = np.moveaxis(np.asarray(data, dtype=float), 0, -1)
counts_flat = counts_cube.reshape(-1, counts_cube.shape[-1])
ratios_flat = counts_to_ratios(counts_flat, transformation="none", nan_filling="none")
ratios_cube = ratios_flat.numpy().reshape(counts_cube.shape)
n_peak = counts_cube.shape[-1]
fig, axes = plt.subplots(2, n_peak, figsize=(4 * n_peak, 7), squeeze=False)
for i, peak in enumerate(peak_names):
c = counts_cube[:, :, i]
r = ratios_cube[:, :, i]
if hide_zero_count:
c = np.where(c == 0, np.nan, c)
if hide_zero_ratio:
r = np.where(r == 0, np.nan, r)
im0 = axes[0, i].imshow(np.log1p(c), cmap="Purples", vmin=0.0)
axes[0, i].set_title(f"Count (log1p)\n{peak}")
axes[0, i].axis("off")
fig.colorbar(im0, ax=axes[0, i], fraction=0.046, pad=0.04)
vmax = np.nanpercentile(ratios_cube, 99) if np.isfinite(ratios_cube).any() else 1.0
im1 = axes[1, i].imshow(r, cmap="Reds", vmin=0.0, vmax=vmax)
axes[1, i].set_title(f"Ratio\n{peak}")
axes[1, i].axis("off")
fig.colorbar(im1, ax=axes[1, i], fraction=0.046, pad=0.04)
fig.suptitle(f"Gene {gene_id} | showing {n_shown}/{n_peaks} peaks | {bin_table}", y=1)
fig.tight_layout()
plt.show()
top_genes = sv_res_16um.head(10)["gene"].astype(str).tolist()
top_genes[:5]
['Septin8', 'Gpm6b', 'Slc8a1', 'Gls', 'Camk1d']
for gene_id in top_genes[:3]:
plot_gene_peak_maps(
sdata=sdata,
bin_table=test_table,
bin_element=test_bins_element,
gene_id=gene_id,
group_col='gene_ids',
max_peaks=6,
peak_meta=peak_meta,
hide_zero_ratio=True,
)
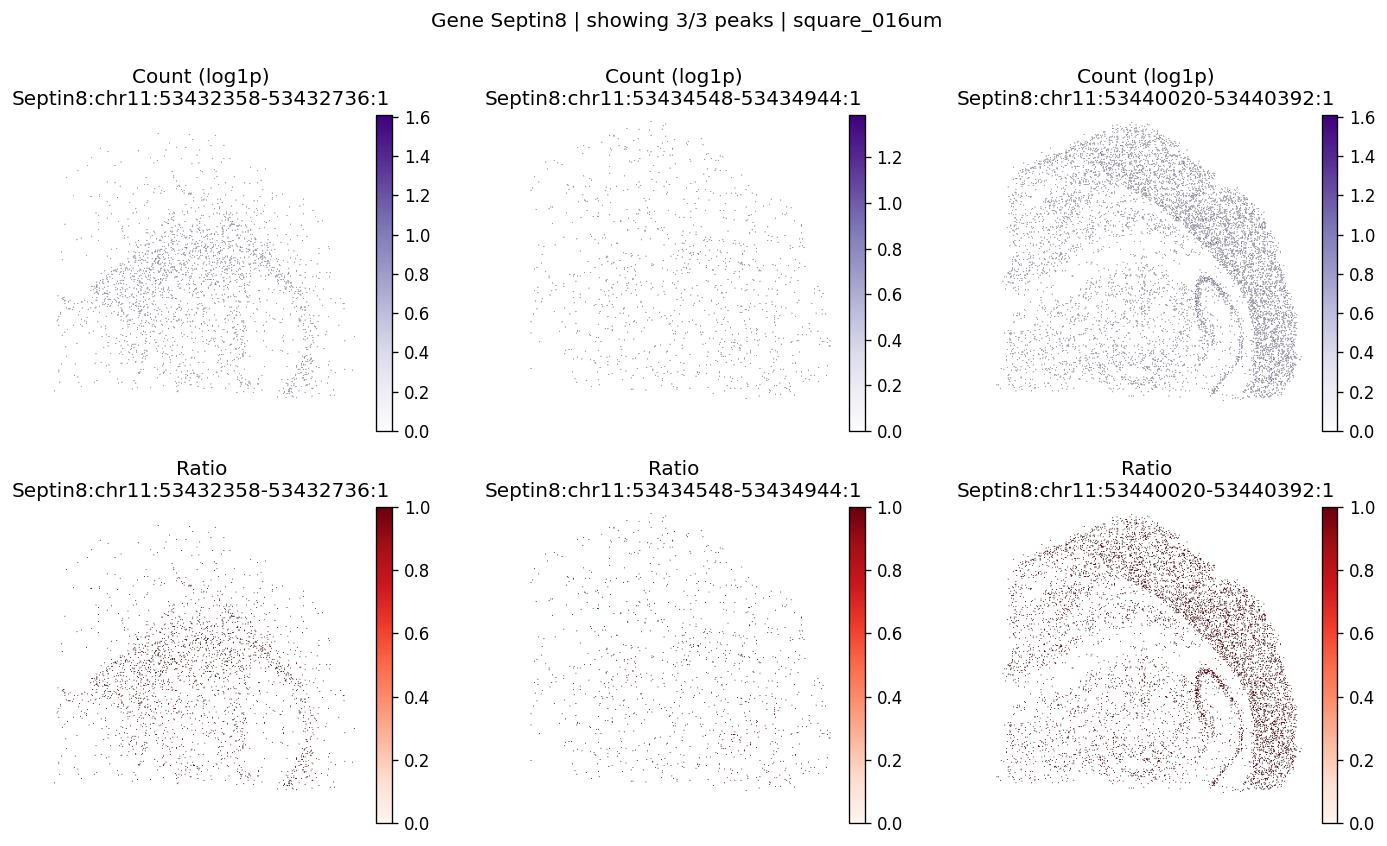
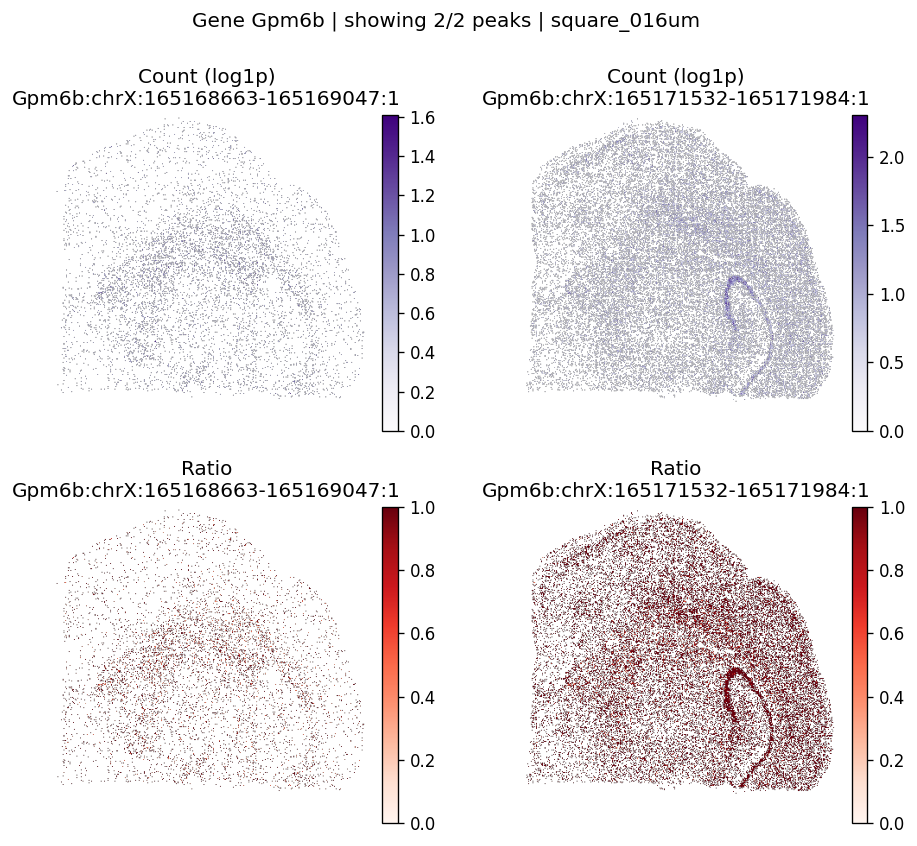
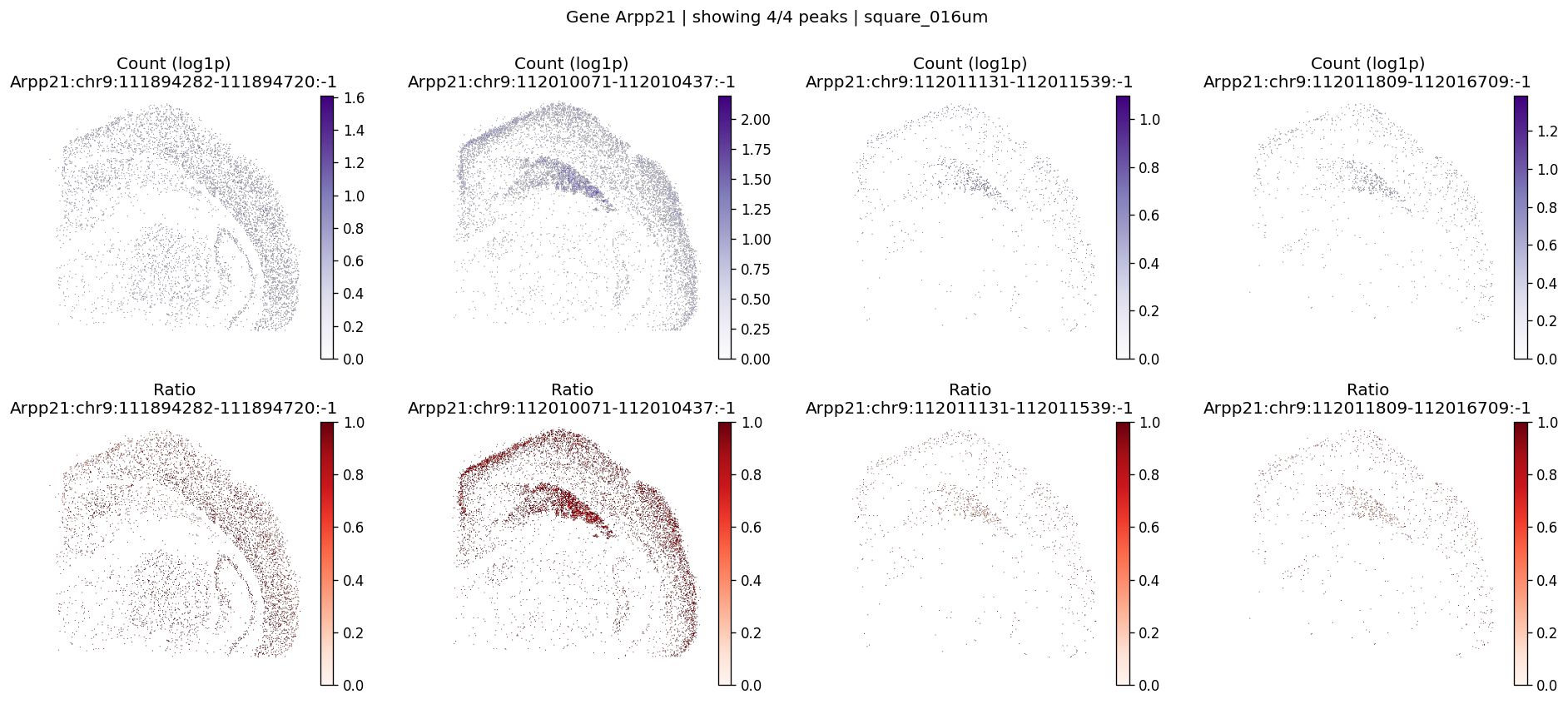
%%time
# Example: inspect a specific gene manually
plot_gene_peak_maps(
sdata, test_table, test_bins_element,
gene_id="Arpp21",
group_col='gene_ids',
peak_meta=peak_meta
)
CPU times: user 256 ms, sys: 31 ms, total: 287 ms
Wall time: 285 ms
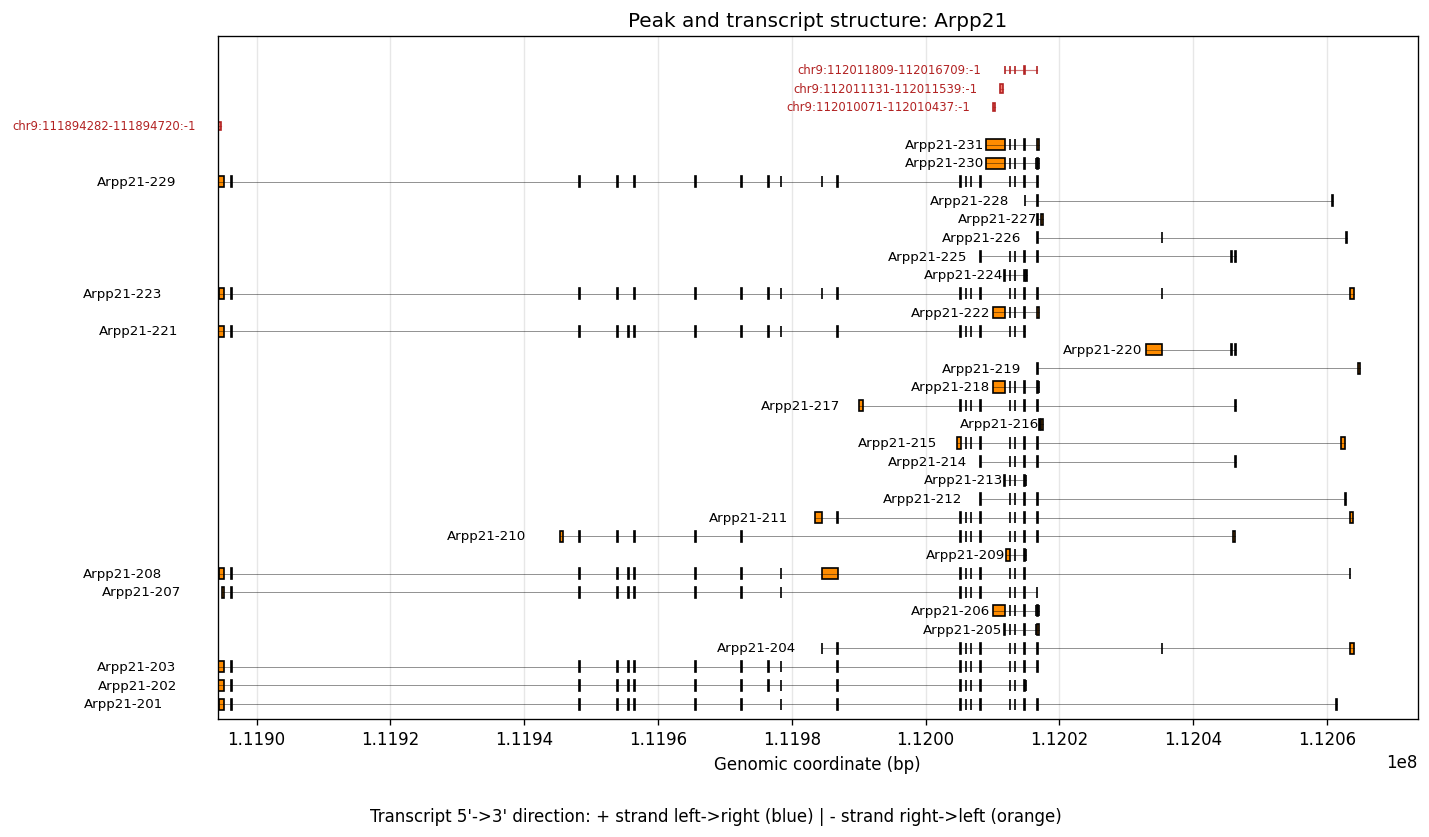
To see the spatially variable transcript regions, we can visualize TREND peaks along with a matched transcript reference. The following files need to be downloaded and provided as input:
peak.txt.bed12from the Sierra peak calling output (seescripts/visiumhd_3p_trend_quant.shfor details)gencode.vM33.annotation.gtf.gzfrom GENCODE
%%time
plot_peak_transcript_structure(
gene_name="Arpp21",
bed12_file=bed12_file,
gtf_file=gtf_file,
)
CPU times: user 4.35 s, sys: 51.6 ms, total: 4.4 s
Wall time: 4.46 s
Advanced analyses#
Spatial resolution comparison#
For illustration, we use the top 200 genes from the 16µm analysis as the reference set:
# np.random.seed(42)
# gene_names = sdata.tables[test_table].var['gene_ids'].unique()
# gene_subset = np.random.choice(gene_names, size=500, replace=False)
gene_subset = sv_res_16um.sort_values('pvalue').head(500)['gene'].astype(str).tolist()
Run spatial variability tests across all three resolutions:
%%time
# Compare results across 2µm, 8µm, 16µm resolutions
resolutions = [
{'bin': f'{dataset_id}_square_002um', 'table_name': 'square_002um'},
{'bin': f'{dataset_id}_square_008um', 'table_name': 'square_008um'},
{'bin': f'{dataset_id}_square_016um', 'table_name': 'square_016um'},
]
res_results = []
for res in resolutions:
m = SplisosmFFT(neighbor_degree=1, rho=0.99)
sdata_subset = sd.filter_by_table_query(
sdata,
table_name=res['table_name'],
var_expr=an.col(gene_name_col).is_in(gene_subset),
)
m.setup_data(
sdata_subset,
bins=res['bin'],
table_name=res['table_name'],
col_key="array_col",
row_key="array_row",
layer="counts",
group_iso_by=group_iso_by,
gene_names=gene_name_col,
min_counts=min_counts,
min_bin_pct=0.0
)
m.test_spatial_variability(method='hsic-ir')
results = m.get_formatted_test_results('sv')[['gene', 'pvalue']].copy()
results.rename(columns={'pvalue': f"p_{res['table_name']}"}, inplace=True)
res_results.append(results)
SV [hsic-ir]: 100%|██████████| 428/428 [04:02<00:00, 1.76it/s]
SV [hsic-ir]: 100%|██████████| 61/61 [00:06<00:00, 8.72it/s]
SV [hsic-ir]: 100%|██████████| 61/61 [00:01<00:00, 33.34it/s]
CPU times: user 12min 47s, sys: 9min 11s, total: 21min 59s
Wall time: 4min 36s
merged = res_results[0]
for res in res_results[1:]:
merged = merged.merge(res, on="gene", how="inner")
p_cols = [c for c in merged.columns if c.startswith("p_")]
print("Spearman correlation across spatial resolutions:")
display(merged[p_cols].corr(method="spearman"))
Spearman correlation across spatial resolutions:
| p_square_002um | p_square_008um | p_square_016um | |
|---|---|---|---|
| p_square_002um | 1.000000 | 0.759201 | 0.629610 |
| p_square_008um | 0.759201 | 1.000000 | 0.901705 |
| p_square_016um | 0.629610 | 0.901705 | 1.000000 |
pairs = list(combinations(p_cols, 2))
if pairs:
fig, axes = plt.subplots(1, len(pairs), figsize=(3 * len(pairs), 3), squeeze=False)
axes = axes.ravel()
for ax, (x_col, y_col) in zip(axes, pairs):
x = merged[x_col].to_numpy()
y = merged[y_col].to_numpy()
corr, pval = spearmanr(x, y)
ax.scatter(x + 1e-300, y + 1e-300, s=8, alpha=0.5)
ax.set_xscale("log")
ax.set_yscale("log")
ax.set_xlabel(x_col)
ax.set_ylabel(y_col)
ax.set_title(f"Spearman ρ={corr:.3f}")
ax.grid(True, alpha=0.3)
low = 1e-300
high = max(np.max(x), np.max(y))
ax.plot([low, high], [low, high], "r--", alpha=0.5, linewidth=1.5)
ax.set_xlim(low, high)
ax.set_ylim(low, high)
plt.tight_layout()
plt.show()
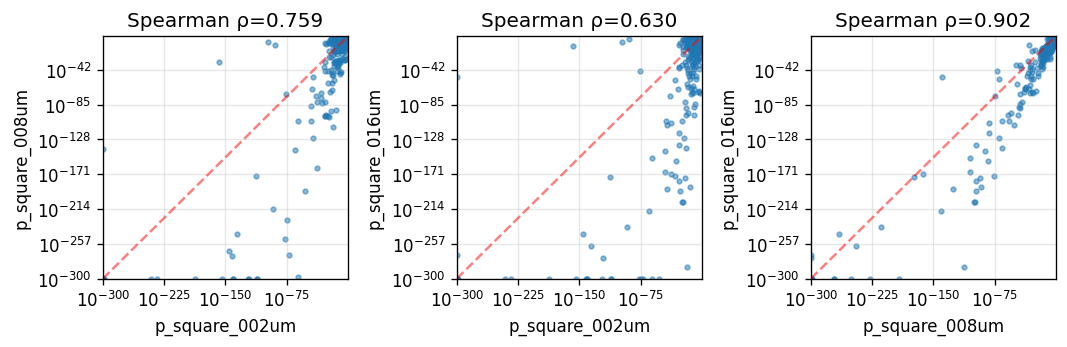
Gene rankings are broadly consistent across resolutions, especially between 16µm and 8µm, with the 16µm analysis showing the strongest statistical significance.
Method comparison: SplisosmFFT vs SplisosmNP#
We now compare FFT-accelerated and non-parametric spatial variability tests at 16 µm resolution.
Note: Since v1.2.0, SplisosmNP SV tests use Liu’s approximation from full-rank spatial-kernel cumulants by default. See the SV hyperparameter optimization notebook for comparison with the legacy low-rank approach.
For large implicit CAR kernels, null_configs={"n_probes": m} controls the Hutchinson trace budget. Smaller m is faster but noisier. To emphasize broad smooth patterns, prefer increasing rho (for example, rho=0.999) rather than rank truncation.
%%time
# Run SplisosmNP at 16µm for direct comparison with SplisosmFFT
model_np = SplisosmNP(
k_neighbors=4,
rho=0.99,
standardize_cov=False, # turn off for faster runtime
)
model_np.setup_data(
adata=sdata.tables[test_table],
spatial_key='spatial', # adata.obsm key for spatial coordinates
layer='counts',
group_iso_by=group_iso_by, # 'gene_ids'
gene_names=gene_name_col, # 'gene_ids'
min_counts=min_counts,
min_bin_pct=min_bin_pct,
filter_single_iso_genes=True,
min_component_size=10 # remove disconnected tissue fragments if any
)
/Users/jysumac/Projects/SPLISOSM/src/splisosm/utils/preprocessing.py:832: UserWarning: Removed 30 spot(s) belonging to graph components with fewer than 10 member(s). 94562 spot(s) remain.
spatial_inputs = _filter_small_components(
CPU times: user 694 ms, sys: 243 ms, total: 937 ms
Wall time: 984 ms
%%time
model_np.test_spatial_variability(
method='hsic-ir',
null_configs={"n_probes": 60},
ratio_transformation='none',
print_progress=True,
)
SV [hsic-ir]: 100%|██████████| 76/76 [00:02<00:00, 29.27it/s]
CPU times: user 22.4 s, sys: 2.3 s, total: 24.6 s
Wall time: 4.45 s
Compare p-values between SplisosmFFT and SplisosmNP:
# Extract results and merge
sv_np = model_np.get_formatted_test_results('sv')[['gene', 'pvalue']].copy()
sv_np = sv_np.rename(columns={'pvalue': 'pvalue_np'})
comparison = sv_res_16um[['gene', 'pvalue']].copy()
comparison = comparison.rename(columns={'pvalue': 'pvalue_fft'})
comparison = comparison.merge(sv_np, on='gene', how='inner')
corr, _ = spearmanr(comparison['pvalue_fft'], comparison['pvalue_np'])
print(f'Genes tested in both methods: {len(comparison)}')
print(f'== Significant in SplisosmFFT (FDR < 0.01): {(comparison["pvalue_fft"] < 0.01).sum()}')
print(f'== Significant in SplisosmNP (FDR < 0.01): {(comparison["pvalue_np"] < 0.01).sum()}')
print(f'== P-value correlation (Spearman rho): {corr:.4f}')
Genes tested in both methods: 1061
== Significant in SplisosmFFT (FDR < 0.01): 540
== Significant in SplisosmNP (FDR < 0.01): 342
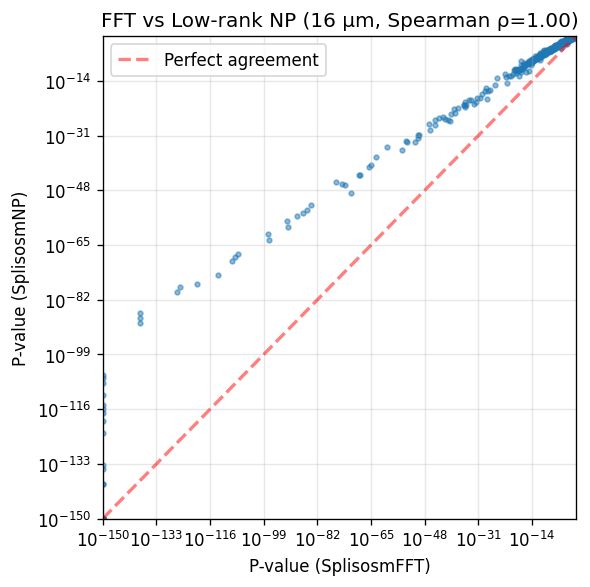
== P-value correlation (Spearman rho): 0.9968
The two approaches yield almost identical rankings. SplisosmFFT shows slightly smaller p-values for top genes due to zero-padding that increases sample size (n=94,592 before vs n=134,688 after rasterization).
# Scatter plot comparison
fig, ax = plt.subplots(figsize=(5, 5))
x = comparison['pvalue_fft'].to_numpy()
y = comparison['pvalue_np'].to_numpy()
ax.scatter(x + 1e-150, y + 1e-150, s=8, alpha=0.5)
ax.set_xscale('log')
ax.set_yscale('log')
ax.set_xlabel('P-value (SplisosmFFT)')
ax.set_ylabel('P-value (SplisosmNP)')
ax.set_title(f'FFT vs Low-rank NP (16 µm, Spearman ρ={corr:.2f})')
ax.grid(True, alpha=0.3)
# Add diagonal reference line
lims = [1e-150, 1.0]
ax.plot(lims, lims, 'r--', alpha=0.5, label='Perfect agreement', linewidth=2)
ax.legend()
ax.set_xlim(lims)
ax.set_ylim(lims)
plt.tight_layout()
plt.show()
Summary and recommendations#
Key findings:
Spatial variability is robustly detectable in this Visium HD 3’ peak-level dataset using
SplisosmFFTon regular grids.Spatial resolution trade-offs:
16 um: Fast and reliable - recommended for initial exploration.
8 um: High agreement with 16 um results.
2 um: Highest resolution but slower and sparser.
SplisosmFFTandSplisosmNPyield concordant rankings on regular grids.
Recommendations:
Start with 16 um binning for exploratory analysis; refine with 8 um if finer spatial detail is needed.
Use SplisosmFFT on regular grids (Visium HD, Xenium binned data) with
neighbor_degree=1, rho=0.99as a robust default; use SplisosmNP for irregular geometries (e.g., cell-segmented data).
For reproducibility#
import sys
from datetime import date
import splisosm
print("Last updated:", date.today())
print("Python:", sys.version.split()[0])
print("splisosm:", getattr(splisosm, "__version__", "unknown"))
Last updated: 2026-05-03
Python: 3.12.13
splisosm: 1.2.0rc1