Welcome#
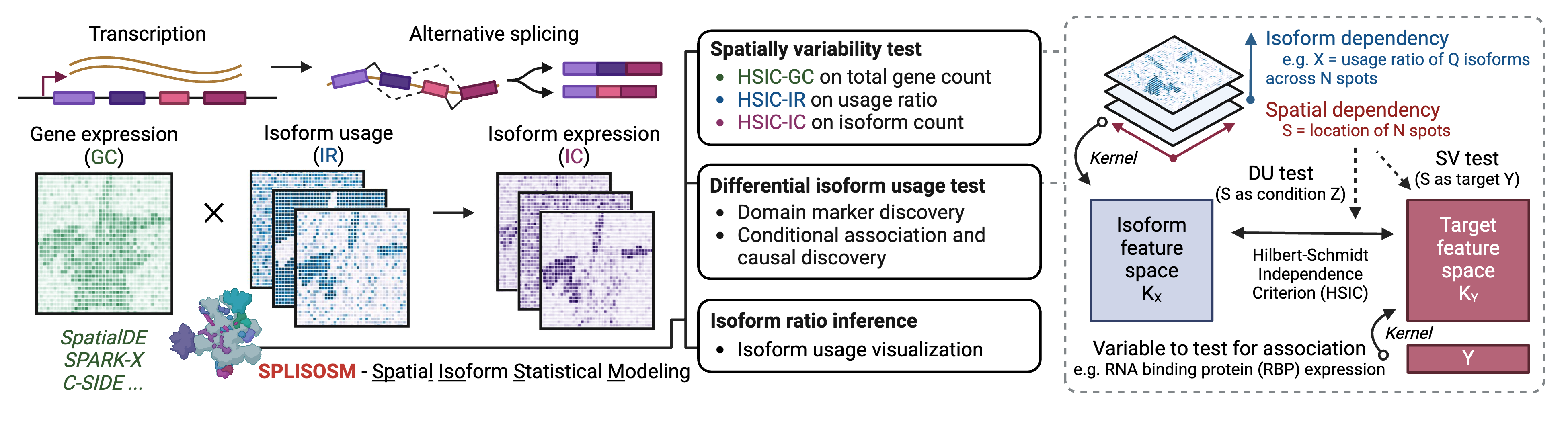
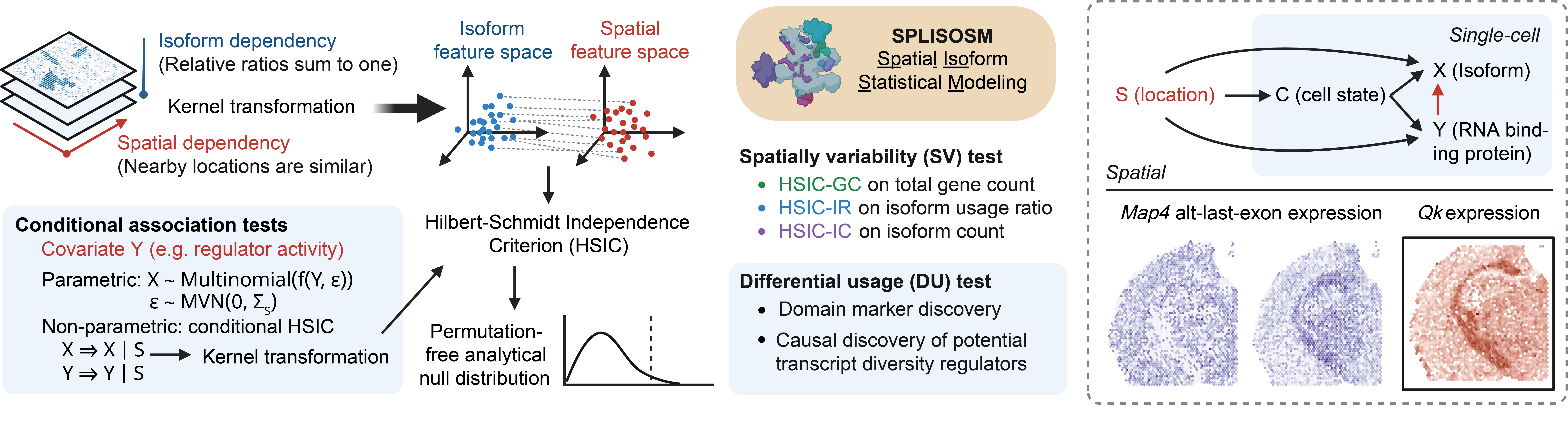
SPLISOSM (SPatiaL ISOform Statistical Modeling) is a Python package for analyzing RNA isoform patterns in spatial transcriptomics (ST) data. It employs multivariate kernel association tests to detect (i) spatial variability in isoform usage across spatial locations, and (ii) differential association between isoform usage and spatial covariates such as region annotation and RNA binding protein (RBP) expression.
Towards isoform-level spatial transcriptomics analysis#
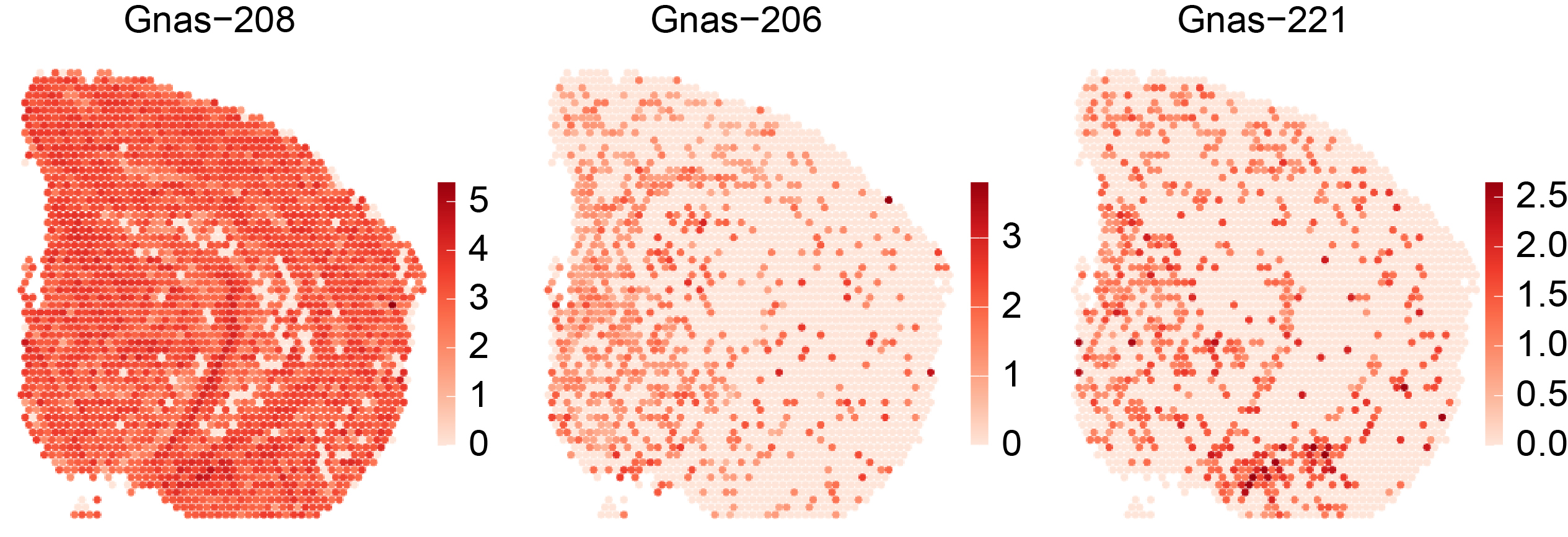
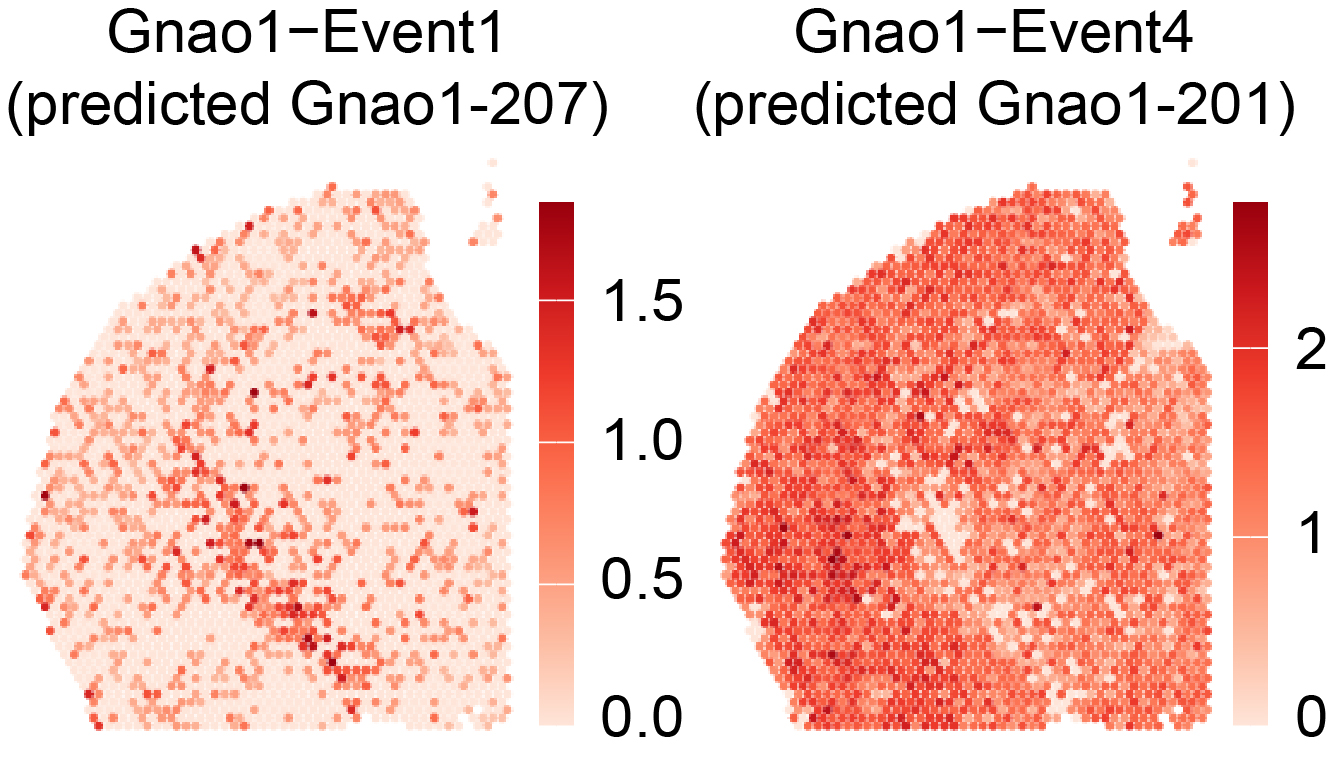
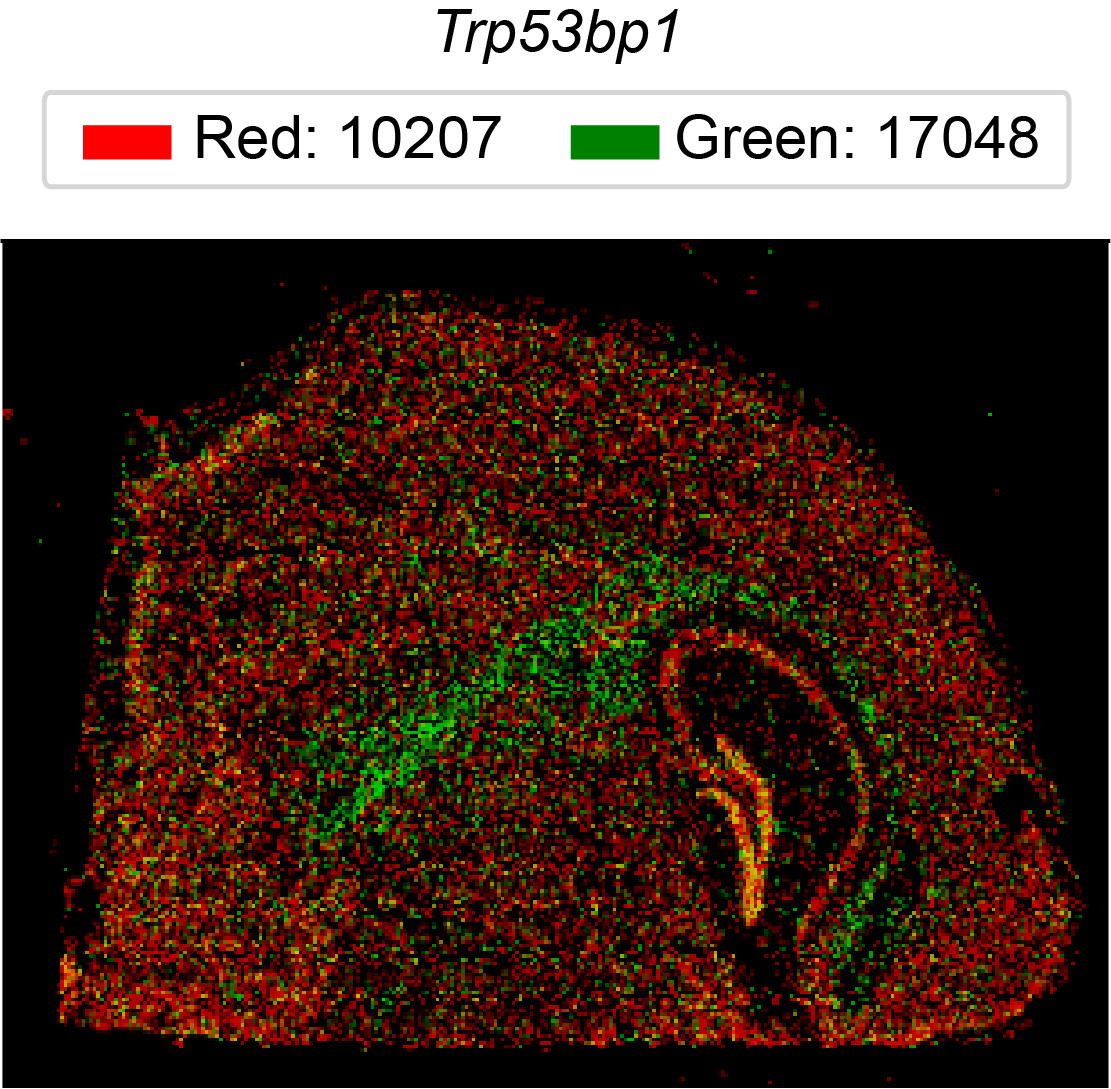
In the associated paper (SPLISOSM 2026) we show that major ST platforms, including short-read-based 10x Visium and imaging-based 10x Xenium Prime 5K, contain sufficient isoform-level information for spatial pattern discovery. These isoform-level signals can be quantified at varying resolutions:
Full-length isoform from long-read data
Transcript 3’ end diversity (TREND) event from short-read data
Exon/junction probe (codeword) usage from imaging-based in situ data
Full-length isoform (long-read) |
TREND event (short-read) |
Codeword (in situ) |
|---|---|---|
|
|
|
Note
SPLISOSM does not perform isoform quantification itself. See the Feature Quantification page for guidance on preparing input data for different platforms.
Unlike conventional gene-centric spatial analysis, SPLISOSM focuses on patterns of RNA processing (e.g., alternative splicing, alternative polyadenylation) manifested at the isoform level. Given isoform quantifications, SPLISOSM supports two types of statistical analyses:
Spatial variability (SV): Detect spatially variable transcript usage (HSIC-IR), transcript expression (HSIC-IC) or gene expression (HSIC-GC). Genes are termed spatially variably processed (SVP) if their HSIC-IR test results are significant; Genes are termed spatially variably expressed (SVE) if their HSIC-GC test results are significant.
Differential isoform usage (DU): Test the conditional association between transcript usage and spatial covariates, such as spatial domains and expression of potential regulators like RBPs.
Note
These tests are multivariate, aggregating signals across all isoforms of a gene. That is, we evaluate whether the joint distribution of isoform quantities varies across space (SV) or is associated with covariates (DU), yielding one test per gene or per gene-covariate pair.
Building on the Hilbert-Schmidt Independence Criterion (HSIC) framework for kernel-based association testing, SPLISOSM achieves provably higher statistical power in sparse data while delivering well-calibrated, permutation-free p-values. For methodological details, please refer to the self-contained Supplementary Notes of the SPLISOSM paper.
Reference#
[SQS+26] Su, Jiayu, et al. “Mapping isoforms and regulatory mechanisms from spatial transcriptomics data with SPLISOSM.” Nature Biotechnology (2026): 1-12. link to paper
Frequently Asked Questions#
Please refer to the FAQ page for answers to common questions regarding installation and usage.
Reporting issues#
If you encounter any issues during installation or usage, please report them on the GitHub Issues page.