Visium HD ONT (isoform)#
This notebook demonstrates a spatial isoform usage workflow for Visium HD with long-read transcript quantification:
Load preprocessed transcript-level
SpatialDatafrom a.zarrstore.Visualize transcript-level count/ratio across space.
Run FFT-accelerated spatial variability tests with
SplisosmFFT.Compare results across spatial resolutions and with
SplisosmNP.
Estimated runtime: ~10 min.
Preliminary notes#
We will use a Visium HD 3’ adult mouse brain data sequenced with Oxford Nanopore generated by EPI2ME. For dataset access and description of the analysis workflow, please check the official page from EPI2ME. Specifically, we downloaded the following outputs from the EPI2ME page:
- spaceranger/hac/HD_Adult_Mouse_Brain/
Space Ranger outputs (`possorted_genome_bam.bam` is not required).
- wf-single-cell/sup/SAMPLE_S1_L001/SAMPLE_S1_L001.transcript_raw_feature_bc_matrix_2um/
transcript-level count matrix (generated by EPI2ME with a custom pipeline).
- wf-single-cell/sup/SAMPLE_S1_L001/SAMPLE_S1_L001.transcriptome.gff.gz
GFF transcript annotation file.
Furthermore, a barcode-by-transcript count matrix raw_tx_bc_matrix.h5 (reference isoforms only) was generated using convert_mtx_to_h5.py, available under the scripts directory.
python convert_mtx_to_h5.py \
'./wf-single-cell/sup/SAMPLE_S1_L001/SAMPLE_S1_L001.transcript_raw_feature_bc_matrix_2um' \
'./wf-single-cell/sup/SAMPLE_S1_L001/SAMPLE_S1_L001.transcriptome.gff.gz' \
'raw_tx_bc_matrix.h5'
Imports#
from __future__ import annotations
import gzip
import re
import warnings
from pathlib import Path
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
from scipy.stats import spearmanr
import spatialdata as sd
import spatialdata_plot
from spatialdata import rasterize_bins
from splisosm import SplisosmFFT, SplisosmNP
from splisosm.utils import counts_to_ratios
/Users/jysumac/miniforge3/envs/splisosm_test/lib/python3.12/site-packages/tqdm/auto.py:21: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html
from .autonotebook import tqdm as notebook_tqdm
warnings.filterwarnings("ignore", category=FutureWarning)
plt.rcParams["figure.dpi"] = 120
plt.rcParams["figure.figsize"] = (6, 4)
Configure paths and core parameters#
# Input data (provided by user)
sdata_zarr = Path("/Users/jysumac/Projects/SPLISOSM_paper/data/visiumhd_ont_mouse_cbs/sdata_tx.filtered.zarr")
gff_file = Path("/Users/jysumac/Projects/SPLISOSM_paper/data/visiumhd_ont_mouse_cbs/SAMPLE_S1_L001.transcriptome.gff.gz")
# Resolution for primary testing
dataset_id = ""
test_table = "square_016um"
test_bins_element = f"{dataset_id}_{test_table}"
# Feature grouping and quality filters
group_iso_candidates = ["gene_ids", "gene_id"]
gene_name_candidates = ["gene_name", "gene_symbol", "gene_names"]
ONT data is generally more sparse than short-read data. To test as many genes as possible, we use a more lenient min_bin_pct threshold (the minimum percentage of non-zero bins required to keep an isoform).
# Isoform and gene filtering thresholds
min_counts = 10
min_bin_pct = 0.001 # a more lenient filter for sparse ONT data
Load preprocessed transcript-level SpatialData#
We now load a preprocessed SpatialData object, generated by load_visiumhd_probe with raw_tx_bc_matrix.h5 as inputs.
%%time
if not sdata_zarr.exists():
# from splisosm.io import load_visiumhd_probe
# sdata = load_visiumhd_probe(
# path="./spaceranger/hac/HD_Adult_Mouse_Brain",
# bin_sizes=[2, 8, 16],
# filtered_counts_file=True,
# path_to_feature_2um_h5="raw_tx_bc_matrix.h5",
# )
# sdata.write_zarr("sdata_tx.filtered.zarr")
raise FileNotFoundError(f"Cannot find Zarr store: {sdata_zarr}")
if not gff_file.exists():
raise FileNotFoundError(f"Cannot find GFF file: {gff_file}")
print("Loading preprocessed SpatialData...")
sdata = sd.read_zarr(sdata_zarr)
sdata
Loading preprocessed SpatialData...
CPU times: user 11 s, sys: 2.84 s, total: 13.8 s
Wall time: 12.1 s
SpatialData object, with associated Zarr store: /Users/jysumac/Projects/SPLISOSM_paper/data/visiumhd_ont_mouse_cbs/sdata_tx.filtered.zarr
├── Images
│ ├── '_hires_image': DataArray[cyx] (3, 3000, 3200)
│ └── '_lowres_image': DataArray[cyx] (3, 563, 600)
├── Shapes
│ ├── '_square_002um': GeoDataFrame shape: (7857218, 1) (2D shapes)
│ ├── '_square_008um': GeoDataFrame shape: (492663, 1) (2D shapes)
│ └── '_square_016um': GeoDataFrame shape: (123658, 1) (2D shapes)
└── Tables
├── 'square_002um': AnnData (7857218, 48001)
├── 'square_008um': AnnData (492663, 48001)
└── 'square_016um': AnnData (123658, 48001)
with coordinate systems:
▸ '', with elements:
_hires_image (Images), _lowres_image (Images), _square_002um (Shapes), _square_008um (Shapes), _square_016um (Shapes)
▸ '_downscaled_hires', with elements:
_hires_image (Images), _square_002um (Shapes), _square_008um (Shapes), _square_016um (Shapes)
▸ '_downscaled_lowres', with elements:
_lowres_image (Images), _square_002um (Shapes), _square_008um (Shapes), _square_016um (Shapes)
Note that cell-segmentation results are not available for this dataset.
def pick_first(columns, candidates):
for c in candidates:
if c in columns:
return c
return None
print("Tables:", sorted(sdata.tables.keys()))
print("Images:", sorted(getattr(sdata, "images", {}).keys()))
print("Shapes:", sorted(getattr(sdata, "shapes", {}).keys()))
if test_table not in sdata.tables:
raise ValueError(f"{test_table} is not available. Choose from: {sorted(sdata.tables.keys())}")
adata_test = sdata.tables[test_table]
group_iso_by = pick_first(adata_test.var.columns, group_iso_candidates)
if group_iso_by is None:
raise ValueError(
"Could not infer grouping column for transcript features. "
f"Available var columns: {list(adata_test.var.columns)}"
)
gene_name_col = pick_first(adata_test.var.columns, gene_name_candidates) or group_iso_by
if test_bins_element not in getattr(sdata, "shapes", {}):
test_bins_element = test_table
print(f"Using table={test_table}, bins={test_bins_element}")
print(f"Grouping column={group_iso_by}, display names={gene_name_col}")
Tables: ['square_002um', 'square_008um', 'square_016um']
Images: ['_hires_image', '_lowres_image']
Shapes: ['_square_002um', '_square_008um', '_square_016um']
Using table=square_016um, bins=_square_016um
Grouping column=gene_ids, display names=gene_name
For each isoform, we have the following metadata columns (inferred from the GFF file):
adata_test.var.head(5)
| gene_ids | probe_ids | feature_types | gene_name | genome | |
|---|---|---|---|---|---|
| ENSMUST00000000001 | ENSMUSG00000000001 | ENSMUST00000000001 | Gene Expression | Gnai3 | |
| ENSMUST00000000028 | ENSMUSG00000000028 | ENSMUST00000000028 | Gene Expression | Cdc45 | |
| ENSMUST00000000033 | ENSMUSG00000048583 | ENSMUST00000000033 | Gene Expression | Igf2 | |
| ENSMUST00000000049 | ENSMUSG00000000049 | ENSMUST00000000049 | Gene Expression | Apoh | |
| ENSMUST00000000058 | ENSMUSG00000000058 | ENSMUST00000000058 | Gene Expression | Cav2 |
As expected, the ONT-based data is sparser than short-read 3’ and targeted Visium HD datasets.
def summarize_table(adata):
X = adata.layers["counts"] if "counts" in adata.layers else adata.X
if hasattr(X, "nnz"):
nnz = int(X.nnz)
total = int(X.shape[0] * X.shape[1])
density = nnz / total if total else np.nan
else:
arr = np.asarray(X)
nnz = int(np.count_nonzero(arr))
total = int(arr.size)
density = nnz / total if total else np.nan
return {
"n_features": int(adata.n_vars),
"n_bins": int(adata.n_obs),
"count_mtx_density": density,
}
rows = []
for key in sorted(sdata.tables.keys()):
if key.startswith("square_"):
rows.append({"table": key, **summarize_table(sdata.tables[key])})
pd.DataFrame(rows).sort_values("table")
| table | n_features | n_bins | count_mtx_density | |
|---|---|---|---|---|
| 0 | square_002um | 48001 | 7857218 | 0.000098 |
| 1 | square_008um | 48001 | 492663 | 0.001379 |
| 2 | square_016um | 48001 | 123658 | 0.004826 |
Finally, take a look at the tissue structure and spatial distribution of a few isoforms.
def ensure_rasterized(sdata, bin_table: str, bin_element: str, layer: str = "counts"):
raster_key = f"rasterized_{bin_table}_{layer}"
if raster_key in sdata.images:
return raster_key
adata = sdata.tables[bin_table]
adata.X = adata.layers[layer] if layer in adata.layers else adata.X
if hasattr(adata.X, "tocsc") and getattr(adata.X, "format", None) != "csc":
adata.X = adata.X.tocsc()
sdata[raster_key] = rasterize_bins(
sdata,
bins=bin_element,
table_name=bin_table,
col_key="array_col",
row_key="array_row",
)
return raster_key
%%time
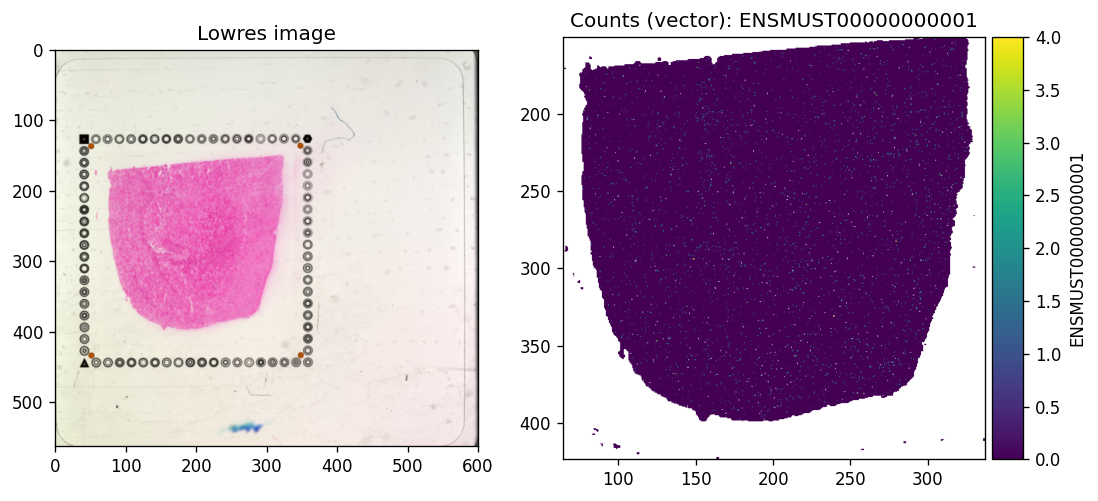
tx_name = 'ENSMUST00000000001' # Gnai3
axes = plt.subplots(1, 2, figsize=(10, 5))[1].flatten()
sdata.pl.render_images(f"{dataset_id}_lowres_image").pl.show(
coordinate_systems=f"{dataset_id}_downscaled_lowres",
ax=axes[0], title="Lowres image"
)
sdata.pl.render_shapes(f"{dataset_id}_square_016um", color=tx_name).pl.show(
coordinate_systems=f"{dataset_id}_downscaled_lowres",
ax=axes[1], title=f"Counts (vector): {tx_name}"
)
Clipping input data to the valid range for imshow with RGB data ([0..1] for floats or [0..255] for integers). Got range [-0.017167382..1.0].
INFO Using 'datashader' backend with 'None' as reduction method to speed up plotting. Depending on the
reduction method, the value range of the plot might change. Set method to 'matplotlib' to disable this
behaviour.
INFO Using the datashader reduction "mean". "max" will give an output very close to the matplotlib result.
CPU times: user 11.7 s, sys: 2.71 s, total: 14.4 s
Wall time: 15.3 s
Spatial variability testing with SplisosmFFT#
model = SplisosmFFT(neighbor_degree=1, rho=0.99)
model.setup_data(
sdata=sdata,
bins=test_bins_element,
table_name=test_table,
col_key="array_col",
row_key="array_row",
layer="counts",
group_iso_by=group_iso_by,
gene_names=gene_name_col,
min_counts=min_counts,
min_bin_pct=min_bin_pct,
)
print(model)
=== FFT SPLISOSM model for spatial isoform testings
- Number of genes: 1558
- Number of observed spots: 123658
- Number of raster cells: 175561
- Average number of isoforms per gene: 2.297175866495507
=== Test results
- Spatial variability test: NA
- Differential usage test: NA
While many genes have more than 10 isoforms detected, the effective number of isoforms (adjusted for expression level) is much smaller.
%%time
gene_meta = model.extract_feature_summary(level="gene")
feature_meta = model.extract_feature_summary(level="isoform")
gene_meta.sort_values("perplexity", ascending=False).head(5)
Genes: 100%|██████████| 1558/1558 [00:01<00:00, 931.17it/s]
CPU times: user 1.71 s, sys: 237 ms, total: 1.95 s
Wall time: 2.16 s
| n_isos | perplexity | pct_bin_on | count_avg | count_std | |
|---|---|---|---|---|---|
| gene | |||||
| Pantr1 | 9 | 7.620990 | 0.024964 | 0.032291 | 0.221045 |
| Mtch1 | 8 | 7.095370 | 0.023144 | 0.029185 | 0.205137 |
| Gas5 | 17 | 6.323551 | 0.111630 | 0.165667 | 0.564609 |
| Egfl7 | 6 | 5.437132 | 0.014726 | 0.018689 | 0.165910 |
| Paxx | 7 | 5.433822 | 0.035930 | 0.048642 | 0.276595 |
Next, we run method="hsic-ir" to detect spatial variability in isoform usage.
%%time
model.test_spatial_variability(
method="hsic-ir",
ratio_transformation="none",
n_jobs=-1,
print_progress=True,
)
SV (hsic-ir): 100%|██████████| 1558/1558 [01:02<00:00, 24.79it/s]
CPU times: user 1min 36s, sys: 18.4 s, total: 1min 54s
Wall time: 1min 3s
sv_res_16um = model.get_formatted_test_results("sv").sort_values("pvalue_adj")
sv_res_16um.merge(
gene_meta.reset_index()[['gene', 'n_isos', 'perplexity', 'pct_bin_on']],
on='gene',
how='left',
).head(5)
| gene | statistic | pvalue | pvalue_adj | n_isos | perplexity | pct_bin_on | |
|---|---|---|---|---|---|---|---|
| 0 | Olfm1 | 7.099970e-07 | 0.0 | 0.0 | 3 | 2.023517 | 0.177611 |
| 1 | Cdc42 | 1.232240e-06 | 0.0 | 0.0 | 2 | 1.990158 | 0.196437 |
| 2 | Rps24 | 1.208481e-06 | 0.0 | 0.0 | 5 | 1.874414 | 0.261601 |
| 3 | Il1rap | 1.729114e-07 | 0.0 | 0.0 | 3 | 2.439950 | 0.025465 |
| 4 | Myl6 | 2.238133e-06 | 0.0 | 0.0 | 6 | 2.569838 | 0.198362 |
Let’s take a look at the top significant genes and their spatial isoform usage patterns.
sig_001 = int((sv_res_16um["pvalue_adj"] < 0.01).sum())
print(
"Spatially variable genes (FDR < 0.01, 16um): "
f"{sig_001} out of {sv_res_16um.shape[0]} total genes"
)
top_genes = sv_res_16um.head(10)["gene"].astype(str).tolist()
top_genes[:5]
Spatially variable genes (FDR < 0.01, 16um): 700 out of 1558 total genes
['Olfm1', 'Cdc42', 'Rps24', 'Il1rap', 'Myl6']
# Visualize maps for a few significant genes
for gene_id in top_genes[:3]:
plot_gene_tx_maps(
sdata=sdata,
bin_table=test_table,
bin_element=test_bins_element,
gene_id=gene_id,
tx_meta=feature_meta,
group_col='gene_name',
max_peaks=6,
hide_zero_ratio=True,
)
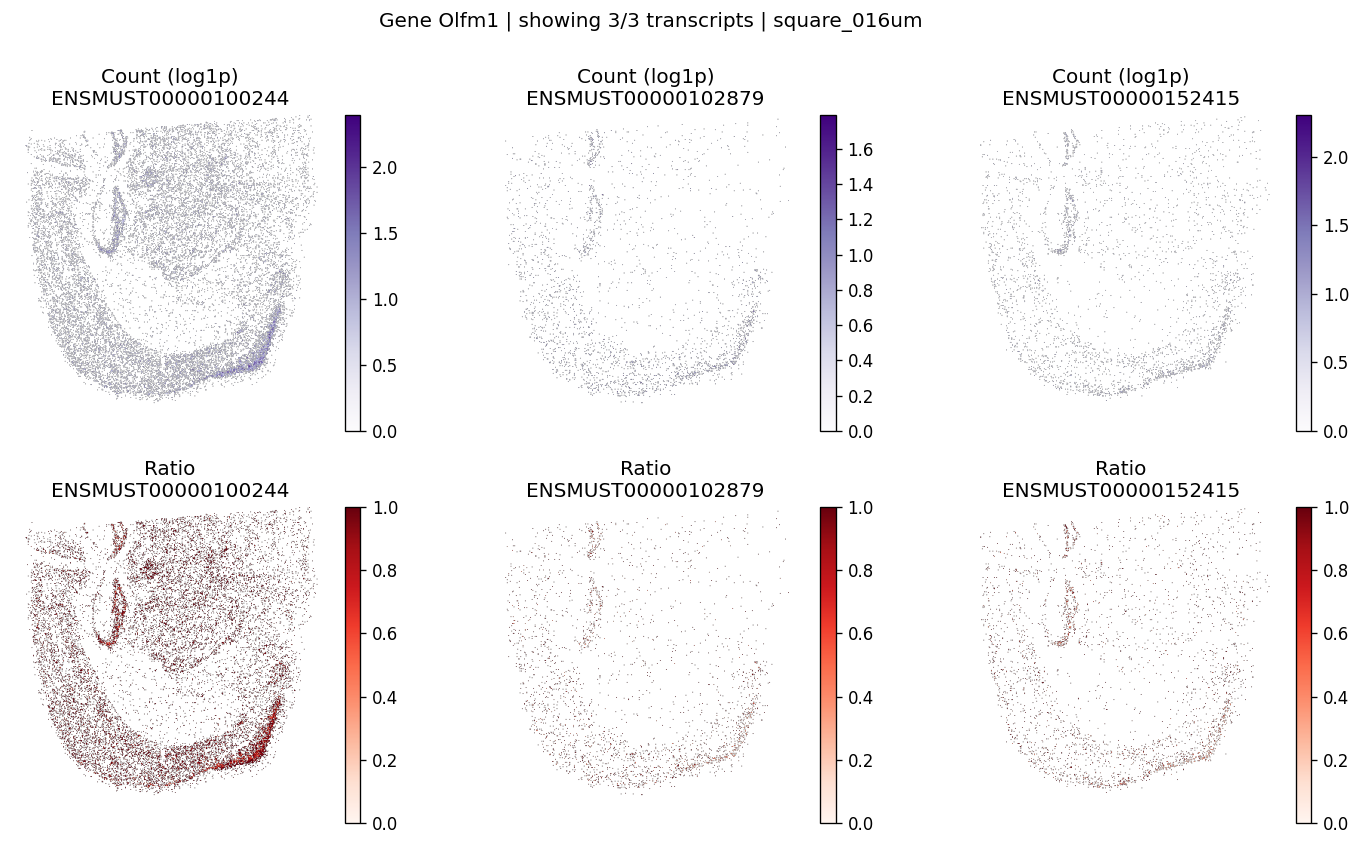
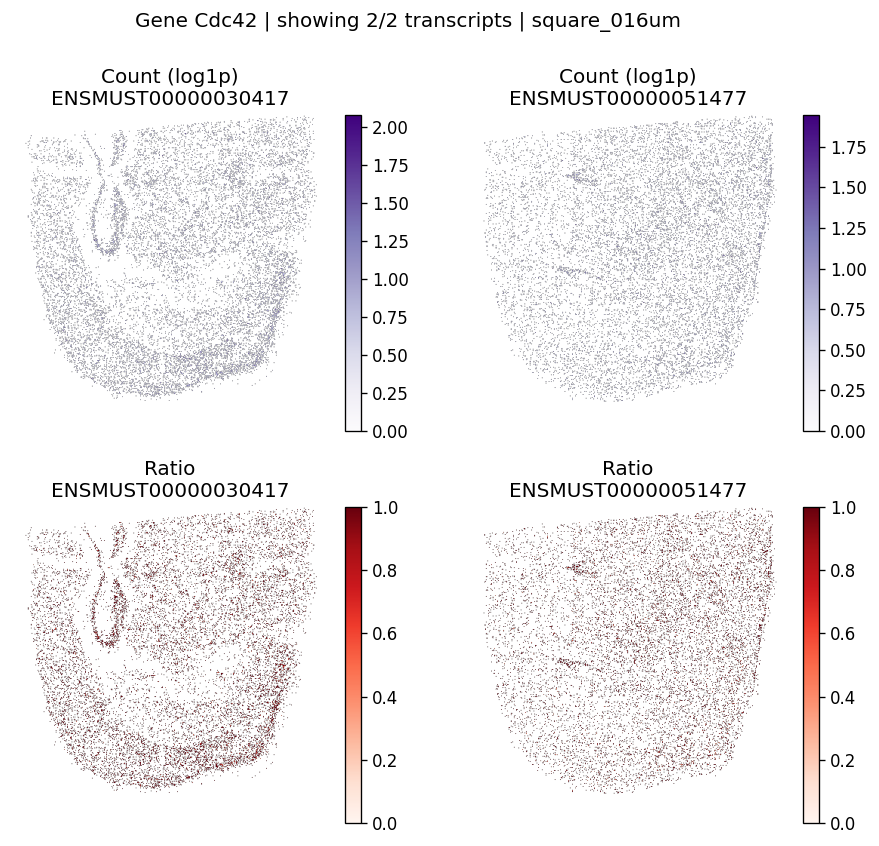
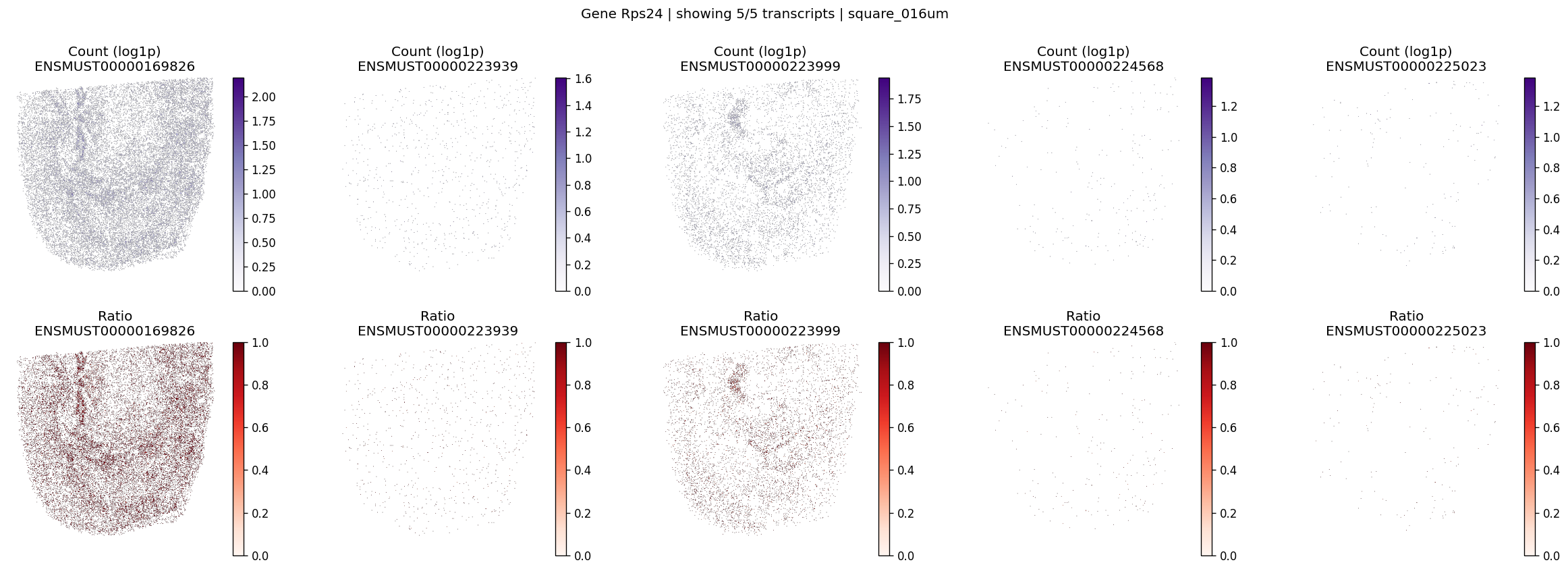
%%time
# Plot an example gene of interest
plot_gene_tx_maps(
sdata=sdata,
bin_table=test_table,
bin_element=test_bins_element,
gene_id='Myl6',
tx_meta=feature_meta,
group_col='gene_name',
max_peaks=6,
hide_zero_ratio=True,
)
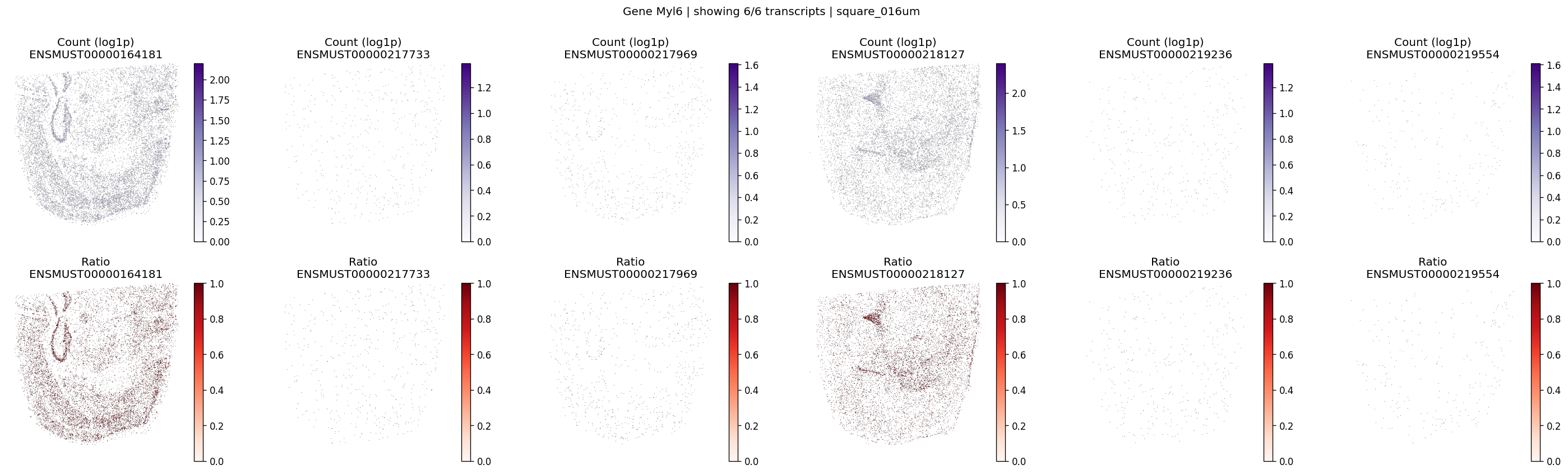
CPU times: user 684 ms, sys: 77.4 ms, total: 761 ms
Wall time: 757 ms
And the transcript structure of a gene (showing kept isoforms after filtering):
%%time
# Plot an example gene of interest with transcript structures
plot_feature_transcript_structure(
sdata=sdata,
bin_table=test_table,
gene_id="Myl6",
feature_meta=feature_meta,
group_col="gene_name",
gff_path=gff_file,
top_k=10,
)
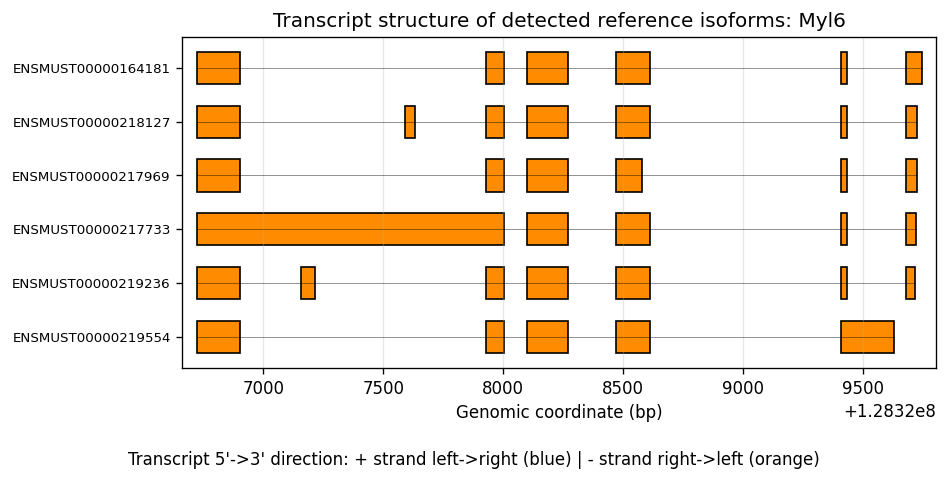
CPU times: user 6.42 s, sys: 252 ms, total: 6.67 s
Wall time: 7.33 s
Comparison with SplisosmNP#
We now compare FFT-accelerated tests with the regular nonparametric tests. SplisosmNP.setup_data performs low-rank kernel approximation (controlled by approx_rank). A higher rank gives more accurate approximations at increased cost. Here we use approx_rank=20 for demonstration.
%%time
# Run SplisosmNP at 16µm for direct comparison with SplisosmFFT
model_np = SplisosmNP()
model_np.setup_data(
adata=sdata.tables[test_table],
spatial_key='spatial', # adata.obsm key for spatial coordinates
layer='counts',
approx_rank=20,
group_iso_by=group_iso_by, # 'gene_ids'
gene_names=gene_name_col, # 'gene_ids'
min_counts=min_counts,
min_bin_pct=min_bin_pct,
)
CPU times: user 5min 13s, sys: 13.4 s, total: 5min 26s
Wall time: 2min 49s
%%time
model_np.test_spatial_variability(
method='hsic-ir',
ratio_transformation='none',
print_progress=True,
)
100%|██████████| 1558/1558 [00:07<00:00, 220.07it/s]
CPU times: user 9.17 s, sys: 1.64 s, total: 10.8 s
Wall time: 7.09 s
Compare p-values between SplisosmFFT and SplisosmNP:
# Extract results and merge
sv_np = model_np.get_formatted_test_results('sv')[['gene', 'pvalue']].copy()
sv_np = sv_np.rename(columns={'pvalue': 'pvalue_np'})
comparison = sv_res_16um[['gene', 'pvalue']].copy()
comparison = comparison.rename(columns={'pvalue': 'pvalue_fft'})
comparison = comparison.merge(sv_np, on='gene', how='inner')
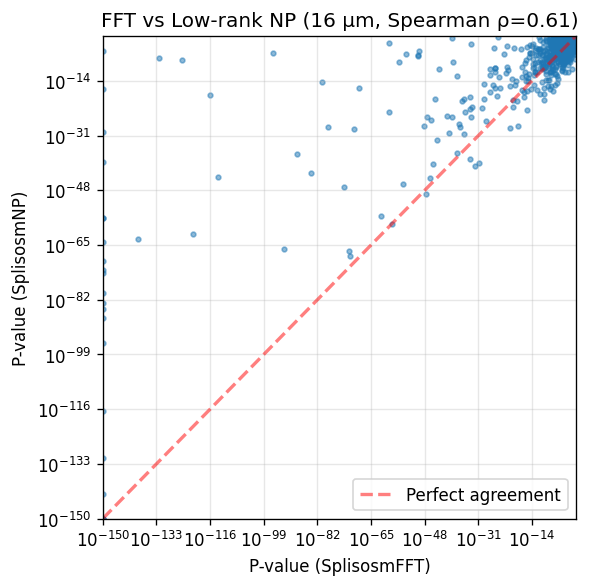
corr, _ = spearmanr(comparison['pvalue_fft'], comparison['pvalue_np'])
print(f'Genes tested in both methods: {len(comparison)}')
print(f'== Significant in SplisosmFFT (FDR < 0.01): {(comparison["pvalue_fft"] < 0.01).sum()}')
print(f'== Significant in SplisosmNP (FDR < 0.01): {(comparison["pvalue_np"] < 0.01).sum()}')
print(f'== P-value correlation (Spearman rho): {corr:.4f}')
Genes tested in both methods: 1558
== Significant in SplisosmFFT (FDR < 0.01): 784
== Significant in SplisosmNP (FDR < 0.01): 625
== P-value correlation (Spearman rho): 0.6145
# Scatter plot comparison
fig, ax = plt.subplots(figsize=(5, 5))
x = comparison['pvalue_fft'].to_numpy()
y = comparison['pvalue_np'].to_numpy()
ax.scatter(x + 1e-150, y + 1e-150, s=8, alpha=0.5)
ax.set_xscale('log')
ax.set_yscale('log')
ax.set_xlabel('P-value (SplisosmFFT)')
ax.set_ylabel('P-value (SplisosmNP)')
ax.set_title(f'FFT vs Low-rank NP (16 µm, Spearman ρ={corr:.2f})')
ax.grid(True, alpha=0.3)
# Add diagonal reference line
lims = [1e-150, 1.0]
ax.plot(lims, lims, 'r--', alpha=0.5, label='Perfect agreement', linewidth=2)
ax.legend()
ax.set_xlim(lims)
ax.set_ylim(lims)
plt.tight_layout()
plt.show()
For reproducibility#
import sys
from datetime import date
import splisosm
print("Last updated:", date.today())
print("Python:", sys.version.split()[0])
print("splisosm:", getattr(splisosm, "__version__", "unknown"))
Last updated: 2026-03-18
Python: 3.12.12
splisosm: 1.0.4