Xenium Prime 5K (codeword)#
This notebook demonstrates a spatial exon/junction variability workflow on Xenium Prime 5K data:
Load codeword-level
SpatialDatausingsplisosm.io.load_xenium_codeword.Inspect binned tables and visualize codeword patterns.
Run FFT-accelerated spatial variability tests with
SplisosmFFT.Compare
SplisosmFFTandSplisosmNPon the same binning grid.
Estimated runtime: ~5 min (excluding SplisosmNP comparison).
Preliminary notes#
For Xenium Prime 5K datasets, each gene is profiled by multiple codewords (i.e., exon/junction probe sets). To extract codeword-level counts, we utilize the load_xenium_codeword function from splisosm.io to read transcripts.zarr.zip directly. The function will aggregate transcript density into multi-resolution square-bin tables and produce a SpatialData object similar to that of the VisiumHD workflow.
Due to Xenium output data structure changes, if you encounter issues, please re-run the Xenium Ranger (>=v3.1.0) relabel pipeline. See 10x documentation for guidance.
Imports#
from __future__ import annotations
from pathlib import Path
import warnings
import numpy as np
import pandas as pd
import matplotlib.pyplot as plt
from scipy.stats import spearmanr
import spatialdata as sd
import spatialdata_plot # Registers plotting accessors
from spatialdata import rasterize_bins
from splisosm import SplisosmFFT, SplisosmNP
from splisosm.utils import counts_to_ratios
from splisosm.io import load_xenium_codeword
/Users/jysumac/miniforge3/envs/splisosm_test/lib/python3.12/site-packages/tqdm/auto.py:21: TqdmWarning: IProgress not found. Please update jupyter and ipywidgets. See https://ipywidgets.readthedocs.io/en/stable/user_install.html
from .autonotebook import tqdm as notebook_tqdm
warnings.filterwarnings('ignore', category=FutureWarning)
plt.rcParams['figure.dpi'] = 120
plt.rcParams['figure.figsize'] = (6, 4)
Configure paths and core parameters#
# Required: Xenium Ranger output directory (either the `outs` dir itself or its parent)
# xenium_prime_outs = Path('/path/to/xenium_prime_5k/outs')
xenium_prime_outs = Path('/Users/jysumac/Projects/SPLISOSM_paper/data/xenium_5k_mouse_brain/codeword-relabel/outs')
# Optional cache path
sdata_zarr = xenium_prime_outs / 'sdata_codeword.filtered.zarr'
# Resolutions (um) to materialize
spatial_resolutions = [8.0, 16.0]
# Primary analysis table / bins
test_table = 'square_016um'
test_bins_element = 'square_016um_bins'
# Feature grouping and filters
group_iso_by = 'gene_symbol'
gene_name_col = 'gene_symbol'
min_counts = 10
min_bin_pct = 0.0
# Xenium transcript filter
quality_threshold = 20.0
Load codeword-level SpatialData#
We use a Xenium Prime 5K mouse brain fresh frozen dataset from 10x Genomics (5K Mouse Pan Tissue and Pathways Panel). Here we use the load_xenium_codeword function to build a SpatialData object where per-codeword density is computed at multi-resolution. The function wraps spatialdata-io.xenium.
%%time
if sdata_zarr.exists():
print('Loading cached SpatialData...')
sdata = sd.read_zarr(sdata_zarr)
else:
print('Building Xenium codeword SpatialData from transcript chunks...')
sdata = load_xenium_codeword(
path=xenium_prime_outs,
spatial_resolutions=spatial_resolutions,
quality_threshold=quality_threshold,
n_jobs=-1,
chunk_batch_size=64,
counts_layer_name='counts',
build_cell_codeword_table=True,
create_square_shapes=True,
show_progress=True,
)
# Optional: cache for faster reruns
# sdata.write(sdata_zarr)
sdata
Building Xenium codeword SpatialData from transcript chunks...
/Users/jysumac/miniforge3/envs/splisosm_test/lib/python3.12/functools.py:912: ImplicitModificationWarning: Transforming to str index.
WARNING The `feature_key` column feature_name is categorical with unknown categories. Please ensure the categories
are known before calling `PointsModel.parse()` to avoid significant performance implications due to the
need for dask to compute the categories. If you did not use PointsModel.parse() explicitly in your code
(e.g. this message is coming from a reader in `spatialdata_io`), please report this finding.
CPU times: user 3min 16s, sys: 57.1 s, total: 4min 13s
Wall time: 2min 13s
SpatialData object
├── Images
│ └── 'morphology_focus': DataTree[cyx] (4, 23912, 34154), (4, 11956, 17077), (4, 5978, 8538), (4, 2989, 4269), (4, 1494, 2134)
├── Labels
│ ├── 'cell_labels': DataTree[yx] (23912, 34154), (11956, 17077), (5978, 8538), (2989, 4269), (1494, 2134)
│ └── 'nucleus_labels': DataTree[yx] (23912, 34154), (11956, 17077), (5978, 8538), (2989, 4269), (1494, 2134)
├── Points
│ └── 'transcripts': DataFrame with shape: (<Delayed>, 13) (3D points)
├── Shapes
│ ├── 'cell_boundaries': GeoDataFrame shape: (63173, 1) (2D shapes)
│ ├── 'nucleus_boundaries': GeoDataFrame shape: (63036, 1) (2D shapes)
│ ├── 'square_008um_bins': GeoDataFrame shape: (576580, 1) (2D shapes)
│ └── 'square_016um_bins': GeoDataFrame shape: (144372, 1) (2D shapes)
└── Tables
├── 'square_008um': AnnData (576580, 11163)
├── 'square_016um': AnnData (144372, 11163)
├── 'table': AnnData (63173, 5006)
└── 'table_codeword': AnnData (63173, 11163)
with coordinate systems:
▸ 'global', with elements:
morphology_focus (Images), cell_labels (Labels), nucleus_labels (Labels), transcripts (Points), cell_boundaries (Shapes), nucleus_boundaries (Shapes), square_008um_bins (Shapes), square_016um_bins (Shapes)
In Xenium Prime 5K datasets, each codeword corresponds to a specific probe set for a gene. Here, we will focus on the codeword-level counts tables:
sdata.tables['table_codeword']contains cell-by-codeword counts aggregated to segmented cells.sdata.tables['table_codeword_square_xxxum']contains bin-by-codeword counts aggregated at various resolutions.
def summarize_table(adata):
X = adata.layers['counts'] if 'counts' in adata.layers else adata.X
if hasattr(X, 'nnz'):
nnz = int(X.nnz)
total = int(X.shape[0] * X.shape[1])
density = nnz / total if total else np.nan
else:
arr = np.asarray(X)
nnz = int(np.count_nonzero(arr))
total = int(arr.size)
density = nnz / total if total else np.nan
return {
'n_features': int(adata.n_vars),
'n_bins': int(adata.n_obs),
'count_mtx_density': density,
}
rows = []
for key in sorted(sdata.tables.keys()):
rows.append({'table': key, **summarize_table(sdata.tables[key])})
table_summary = pd.DataFrame(rows).sort_values('table')
table_summary
| table | n_features | n_bins | count_mtx_density | |
|---|---|---|---|---|
| 0 | square_008um | 11163 | 576580 | 0.018701 |
| 1 | square_016um | 11163 | 144372 | 0.058018 |
| 2 | table | 5006 | 63173 | 0.139117 |
| 3 | table_codeword | 11163 | 63173 | 0.077513 |
Note that only data with regular spacing (e.g., square_016um) are suitable for FFT-based spatial variability analysis.
For illustration purposes, we will run analysis on the square_016um table and use the shape square_016um_bins (i.e., spatial grid with 16x16um bins) for rasterization.
print('Tables:', sorted(sdata.tables.keys()))
print('Shapes:', sorted(getattr(sdata, 'shapes', {}).keys()))
print('Images:', sorted(getattr(sdata, 'images', {}).keys()))
if test_table not in sdata.tables:
raise ValueError(f'{test_table} is not available. Choose from: {sorted(sdata.tables.keys())}')
if test_bins_element not in sdata.shapes:
raise ValueError(f'{test_bins_element} is not available. Choose from: {sorted(sdata.shapes.keys())}')
adata_test = sdata.tables[test_table]
if group_iso_by not in adata_test.var.columns:
raise ValueError(f'{group_iso_by} not found in {test_table}.var columns')
print(f'Using table={test_table}, bins={test_bins_element}')
print(f'Grouping column={group_iso_by}, display names={gene_name_col}')
Tables: ['square_008um', 'square_016um', 'table', 'table_codeword']
Shapes: ['cell_boundaries', 'nucleus_boundaries', 'square_008um_bins', 'square_016um_bins']
Images: ['morphology_focus']
Using table=square_016um, bins=square_016um_bins
Grouping column=gene_symbol, display names=gene_symbol
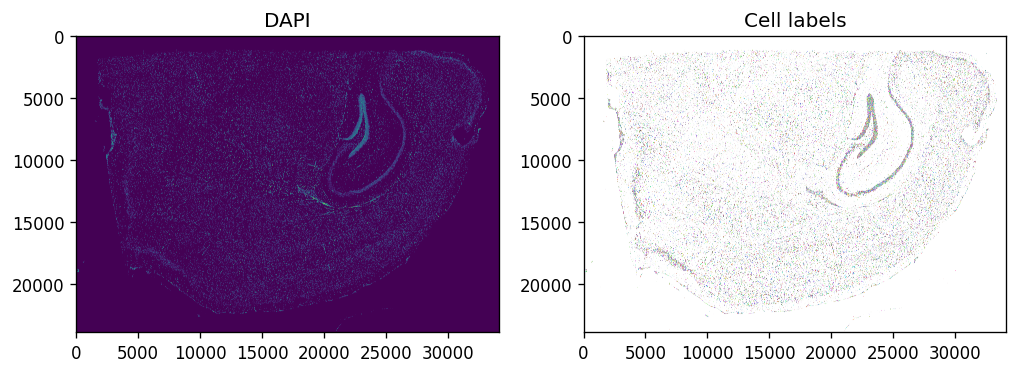
Optional morphology and segmentation preview#
%%time
axes = plt.subplots(1, 2, figsize=(10, 5))[1].flatten()
sdata.pl.render_images(f"morphology_focus", channel='DAPI').pl.show(
coordinate_systems=f"global",
ax=axes[0], title="DAPI", colorbar=False
)
sdata.pl.render_labels(f"cell_labels").pl.show(
coordinate_systems=f"global",
ax=axes[1], title="Cell labels"
)
INFO Rasterizing image for faster rendering.
/Users/jysumac/miniforge3/envs/splisosm_test/lib/python3.12/site-packages/pims/tiff_stack.py:131: UserWarning: <tifffile.TiffPage 0 @16> reading array from closed file
INFO Rasterizing image for faster rendering.
CPU times: user 4min 45s, sys: 23.2 s, total: 5min 8s
Wall time: 1min 4s
Rasterize bins and visualize one codeword#
%%time
# rasterize_bins() expects a CSC matrix for best compatibility
adata_plot = sdata.tables[test_table]
adata_plot.X = adata_plot.layers['counts']
if hasattr(adata_plot.X, 'tocsc') and getattr(adata_plot.X, 'format', None) != 'csc':
adata_plot.X = adata_plot.X.tocsc()
raster_key = f'rasterized_{test_table}'
sdata[raster_key] = rasterize_bins(
sdata,
bins=test_bins_element,
table_name=test_table,
col_key='array_col',
row_key='array_row',
)
print('Created:', raster_key)
Created: rasterized_square_016um
CPU times: user 1.22 s, sys: 416 ms, total: 1.63 s
Wall time: 2.91 s
%%time
feature_name = 'Trp53bp1|17048' # Trp53bp1
img = np.asarray(sdata[f'rasterized_{test_table}'].sel(c=feature_name).values).squeeze()
fig, ax = plt.subplots(figsize=(5, 4))
im = ax.imshow(np.log1p(img), cmap='magma')
ax.set_title(f'{feature_name} (log1p rasterized counts)')
ax.axis('off')
fig.colorbar(im, ax=ax, fraction=0.046, pad=0.04)
plt.show()

CPU times: user 59.1 ms, sys: 11.9 ms, total: 70.9 ms
Wall time: 80.4 ms
Spatial variability testing with SplisosmFFT#
model = SplisosmFFT(neighbor_degree=1, rho=0.99)
model.setup_data(
sdata=sdata,
bins=test_bins_element,
table_name=test_table,
col_key='array_col',
row_key='array_row',
layer='counts',
group_iso_by=group_iso_by,
gene_names=gene_name_col,
min_counts=min_counts,
min_bin_pct=min_bin_pct,
)
print(model)
=== FFT SPLISOSM model for spatial isoform testings
- Number of genes: 4972
- Number of observed spots: 144372
- Number of raster cells: 144372
- Average number of isoforms per gene: 2.091713596138375
=== Test results
- Spatial variability test: NA
- Differential usage test: NA
Before running the test, we can again check gene-level summaries to confirm that Xenium Prime 5K data contains multiple codewords per gene.
%%time
gene_meta = model.extract_feature_summary(level='gene')
gene_meta.sort_values('perplexity', ascending=False).head(5)
Genes: 100%|██████████| 4972/4972 [00:11<00:00, 433.17it/s]
CPU times: user 10.3 s, sys: 1.59 s, total: 11.9 s
Wall time: 12 s
| n_isos | perplexity | pct_bin_on | count_avg | count_std | |
|---|---|---|---|---|---|
| gene | |||||
| Gnao1 | 4 | 3.979105 | 0.616671 | 4.027173 | 5.066839 |
| Apob | 4 | 3.952770 | 0.001385 | 0.002016 | 0.141543 |
| Gja1 | 4 | 3.929913 | 0.375149 | 1.565989 | 3.772387 |
| Acox1 | 4 | 3.921495 | 0.459334 | 1.000139 | 1.440174 |
| Ghr | 4 | 3.909349 | 0.041268 | 0.045805 | 0.233034 |
%%time
model.test_spatial_variability(
method='hsic-ir',
ratio_transformation='none',
n_jobs=-1,
print_progress=True,
)
sv_res_fft = model.get_formatted_test_results('sv').sort_values('pvalue_adj')
SV (hsic-ir): 100%|██████████| 4972/4972 [01:10<00:00, 70.61it/s]
CPU times: user 2min 40s, sys: 33.9 s, total: 3min 14s
Wall time: 1min 10s
sig_001 = int((sv_res_fft['pvalue_adj'] < 0.01).sum())
print(
'Spatially variable genes (FDR < 0.01): '
f'{sig_001} out of {sv_res_fft.shape[0]} total genes'
)
sv_res_fft.head(5)
Spatially variable genes (FDR < 0.01): 2144 out of 4972 total genes
| gene | statistic | pvalue | pvalue_adj | |
|---|---|---|---|---|
| 2061 | Zfp281 | 1.777472e-06 | 0.0 | 0.0 |
| 2310 | Ddit3 | 1.621363e-06 | 0.0 | 0.0 |
| 3851 | Ttll6 | 2.199914e-08 | 0.0 | 0.0 |
| 138 | Grpel1 | 5.033253e-07 | 0.0 | 0.0 |
| 4577 | Ifit3 | 4.637240e-07 | 0.0 | 0.0 |
We can use negative control probes to estimate false positive rates due to technical noise (e.g., probe malfunction or non-specific hybridization).
ctrl_genes = (
sv_res_fft['gene'].str.startswith('NegControlProbe') |
sv_res_fft['gene'].str.startswith('Intergenic')
)
sig_001_ctrl = int((sv_res_fft.loc[ctrl_genes, 'pvalue_adj'] < 0.01).sum())
print(
'Spatially variable negative control genes (FDR < 0.01): '
f'{sig_001_ctrl} out of {sv_res_fft.loc[ctrl_genes].shape[0]} total control genes'
)
sv_res_fft.loc[ctrl_genes].sort_values('pvalue_adj').head(5)
Spatially variable negative control genes (FDR < 0.01): 8 out of 57 total control genes
| gene | statistic | pvalue | pvalue_adj | |
|---|---|---|---|---|
| 2475 | Intergenic_Region_105_part_73 | 2.199321e-08 | 8.115485e-17 | 5.942591e-16 |
| 463 | NegControlProbe_00041 | 5.502025e-09 | 1.810361e-07 | 7.667050e-07 |
| 731 | NegControlProbe_00003 | 5.328555e-09 | 3.139711e-07 | 1.294415e-06 |
| 1042 | Intergenic_Region_10757_part_30 | 8.702548e-09 | 9.215231e-05 | 2.883457e-04 |
| 1105 | NegControlProbe_00014 | 1.269646e-08 | 2.698407e-04 | 7.892047e-04 |
Visualize selected significant genes#
def ensure_rasterized(sdata, bin_table: str, bin_element: str, layer: str = 'counts'):
raster_key = f'rasterized_{bin_table}_{layer}'
if raster_key in sdata.images:
return raster_key
adata = sdata.tables[bin_table]
adata.X = adata.layers[layer]
if hasattr(adata.X, 'tocsc') and getattr(adata.X, 'format', None) != 'csc':
adata.X = adata.X.tocsc()
sdata[raster_key] = rasterize_bins(
sdata,
bins=bin_element,
table_name=bin_table,
col_key='array_col',
row_key='array_row',
)
return raster_key
def plot_gene_codeword_maps(
sdata,
bin_table: str,
bin_element: str,
gene_id: str,
var_meta: pd.DataFrame | None = None,
group_col: str = 'gene_symbol',
max_features: int = 4,
hide_zero_count: bool = True,
hide_zero_ratio: bool = True,
):
adata = sdata.tables[bin_table]
if var_meta is None:
var_meta = adata.var.copy()
if group_col not in var_meta.columns:
raise ValueError(f"'{group_col}' not found in var metadata columns")
feature_names = var_meta.index[var_meta[group_col].astype(str) == str(gene_id)].tolist()
if len(feature_names) == 0:
raise ValueError(f"No features found for gene '{gene_id}'")
feature_names = feature_names[: min(len(feature_names), max_features)]
raster_key = ensure_rasterized(sdata, bin_table=bin_table, bin_element=bin_element)
data = sdata[raster_key].sel(c=feature_names).values
counts_cube = np.moveaxis(np.asarray(data, dtype=float), 0, -1)
counts_flat = counts_cube.reshape(-1, counts_cube.shape[-1])
ratios_flat = counts_to_ratios(counts_flat, transformation='none', nan_filling='none')
ratios_cube = ratios_flat.numpy().reshape(counts_cube.shape)
n_feat = counts_cube.shape[-1]
fig, axes = plt.subplots(2, n_feat, figsize=(4 * n_feat, 7), squeeze=False)
vmax_ratio = np.nanpercentile(ratios_cube, 99) if np.isfinite(ratios_cube).any() else 1.0
for i, feature in enumerate(feature_names):
c = counts_cube[:, :, i]
r = ratios_cube[:, :, i]
if hide_zero_count:
c = np.where(c == 0, np.nan, c)
if hide_zero_ratio:
r = np.where(r == 0, np.nan, r)
im0 = axes[0, i].imshow(np.log1p(c), cmap='Purples', vmin=0.0)
axes[0, i].set_title(f'Count (log1p)\n{feature}')
axes[0, i].axis('off')
fig.colorbar(im0, ax=axes[0, i], fraction=0.046, pad=0.04)
im1 = axes[1, i].imshow(r, cmap='Reds', vmin=0.0, vmax=vmax_ratio)
axes[1, i].set_title(f'Ratio\n{feature}')
axes[1, i].axis('off')
fig.colorbar(im1, ax=axes[1, i], fraction=0.046, pad=0.04)
fig.suptitle(f'Gene {gene_id} | showing {n_feat} features | {bin_table}', y=1.02)
fig.tight_layout()
plt.show()
top_genes = sv_res_fft.head(10)['gene'].astype(str).tolist()
top_genes[:5]
['Zfp281', 'Ddit3', 'Ttll6', 'Grpel1', 'Ifit3']
for gene_id in top_genes[:2]:
plot_gene_codeword_maps(
sdata=sdata,
bin_table=test_table,
bin_element=test_bins_element,
gene_id=gene_id,
var_meta=sdata.tables[test_table].var,
group_col=group_iso_by,
max_features=6,
hide_zero_ratio=True,
)
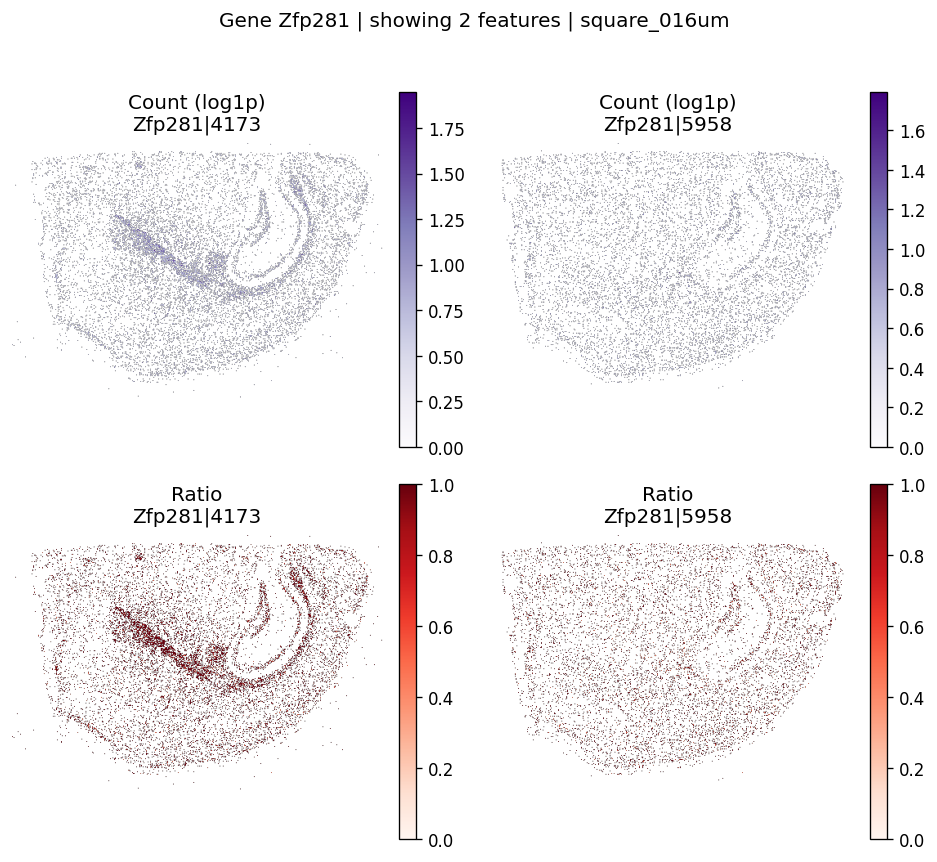
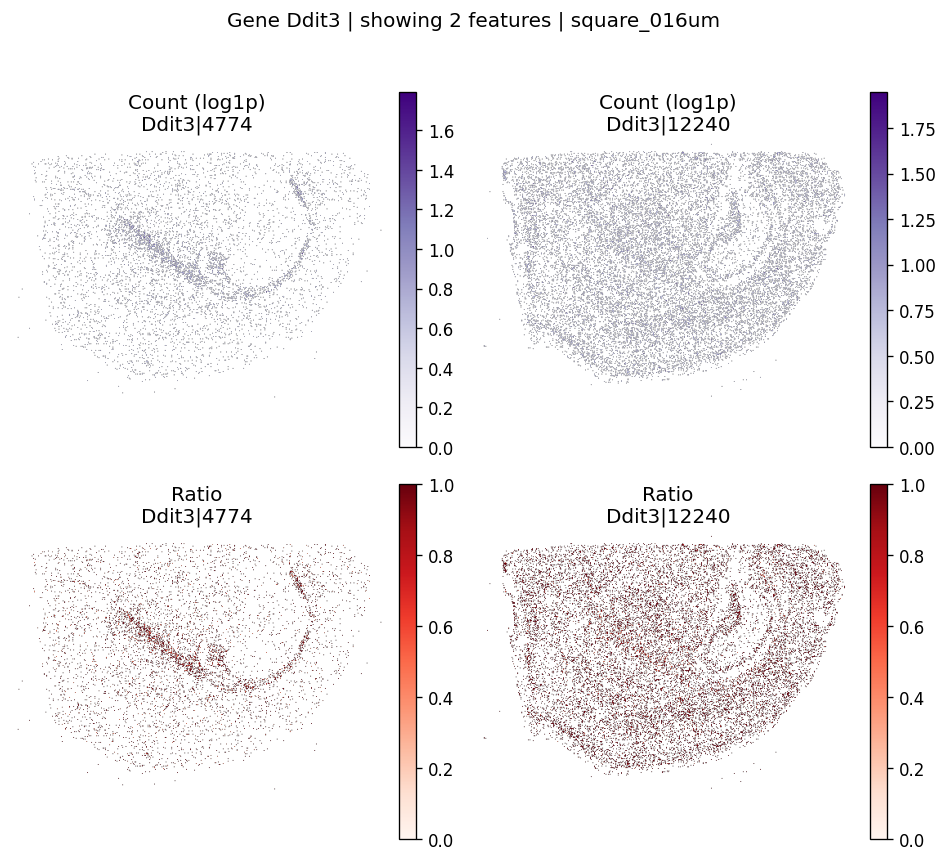
And also for negative control probes
top_ctrl_genes = sv_res_fft.loc[ctrl_genes].head(10)['gene'].astype(str).tolist()
top_ctrl_genes[:5]
['Intergenic_Region_105_part_73',
'NegControlProbe_00041',
'NegControlProbe_00003',
'Intergenic_Region_10757_part_30',
'NegControlProbe_00014']
for gene_id in top_ctrl_genes[:2]:
plot_gene_codeword_maps(
sdata=sdata,
bin_table=test_table,
bin_element=test_bins_element,
gene_id=gene_id,
var_meta=sdata.tables[test_table].var,
group_col=group_iso_by,
max_features=6,
hide_zero_ratio=True,
)
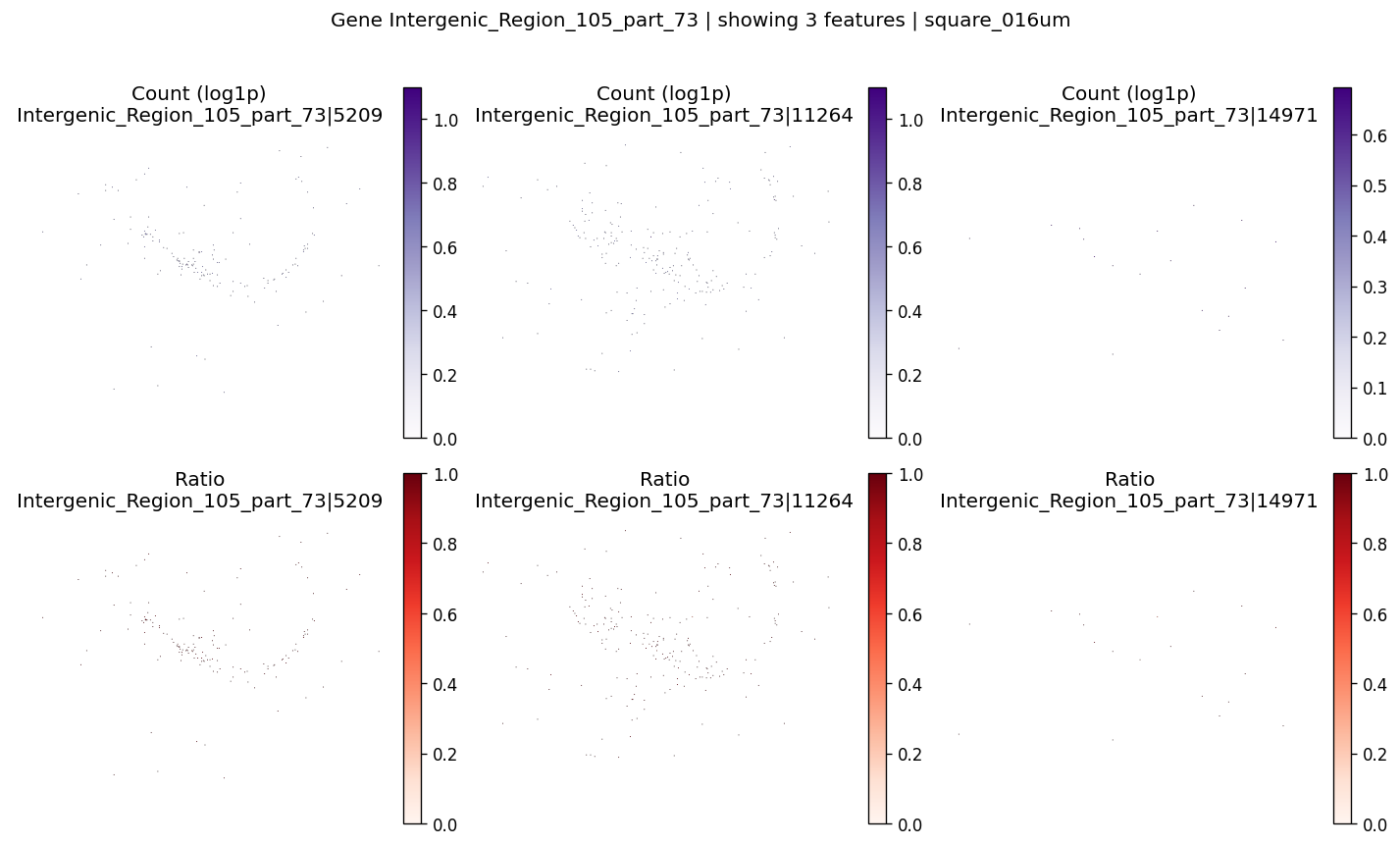
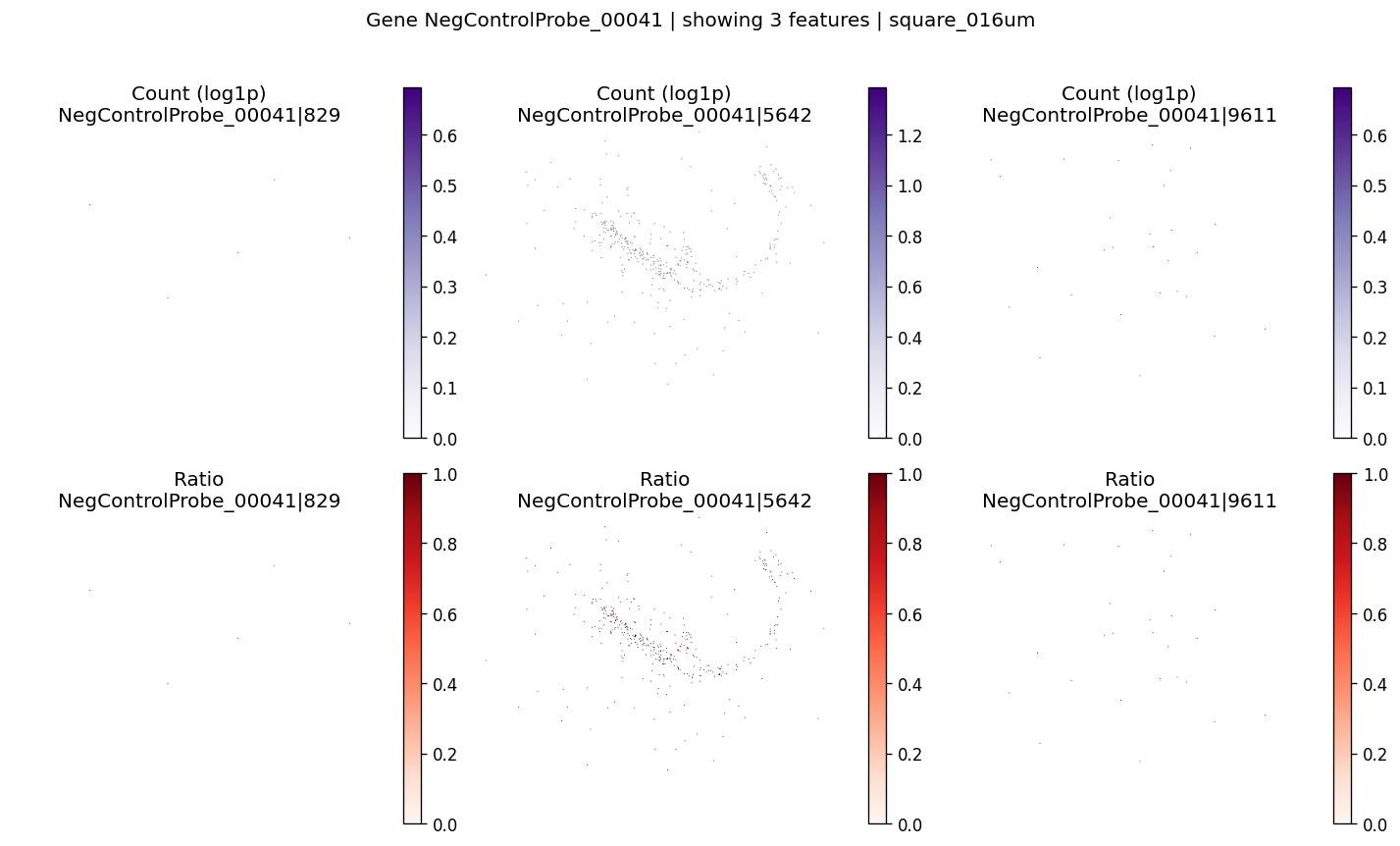
%%time
# Example: inspect a specific gene manually
plot_gene_codeword_maps(
sdata=sdata,
bin_table=test_table,
bin_element=test_bins_element,
gene_id='Dtnbp1',
var_meta=sdata.tables[test_table].var,
group_col=group_iso_by,
max_features=6,
hide_zero_ratio=True,
)
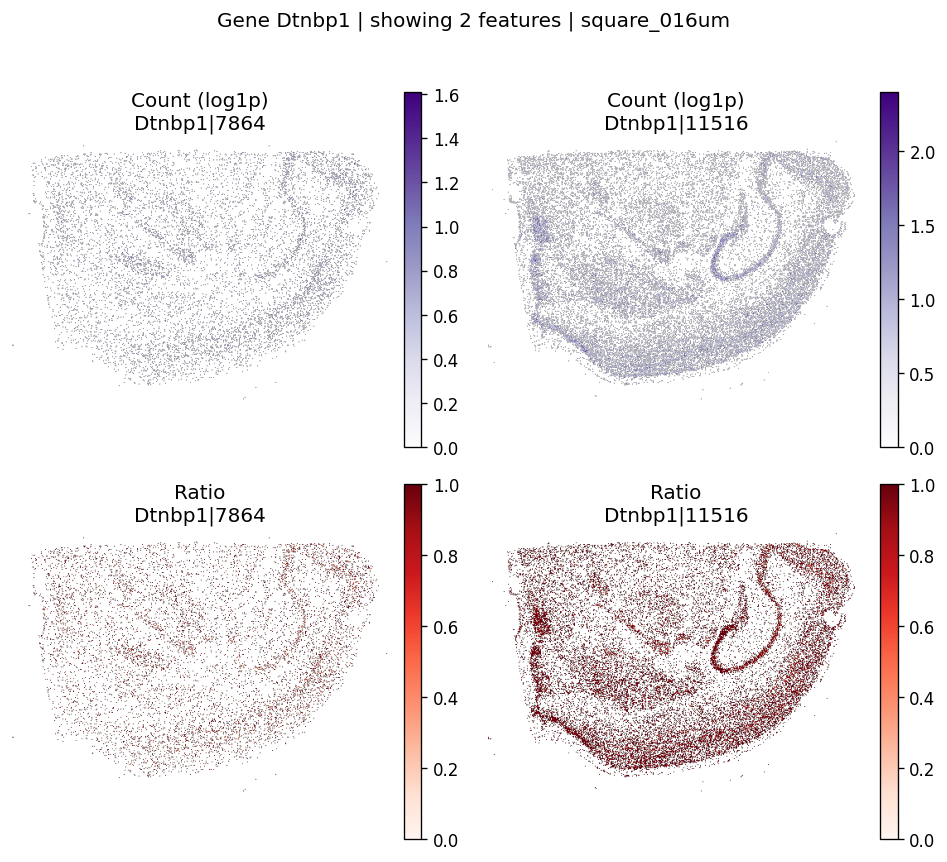
CPU times: user 222 ms, sys: 23.5 ms, total: 246 ms
Wall time: 246 ms
To see the spatially variable transcript regions, we can visualize the 10x reference probe sets along with a matched transcript reference. The following files need to be downloaded and provided as input:
xenium_mouse_5K_gene_expression_panel_probe_locations.bedfrom 10x Genomicsgencode.vM23.annotation.gtf.gzfrom GENCODE
%%time
plot_codeword_transcript_structure(
gene_name="Dtnbp1",
bed_file="/Users/jysumac/Projects/SPLISOSM_paper/data/xenium_5k_mouse_brain/xenium_mouse_5K_gene_expression_panel_probe_locations.bed",
gtf_file="/Users/jysumac/reference/mm10/gencode.vM23.annotation.gtf.gz",
)
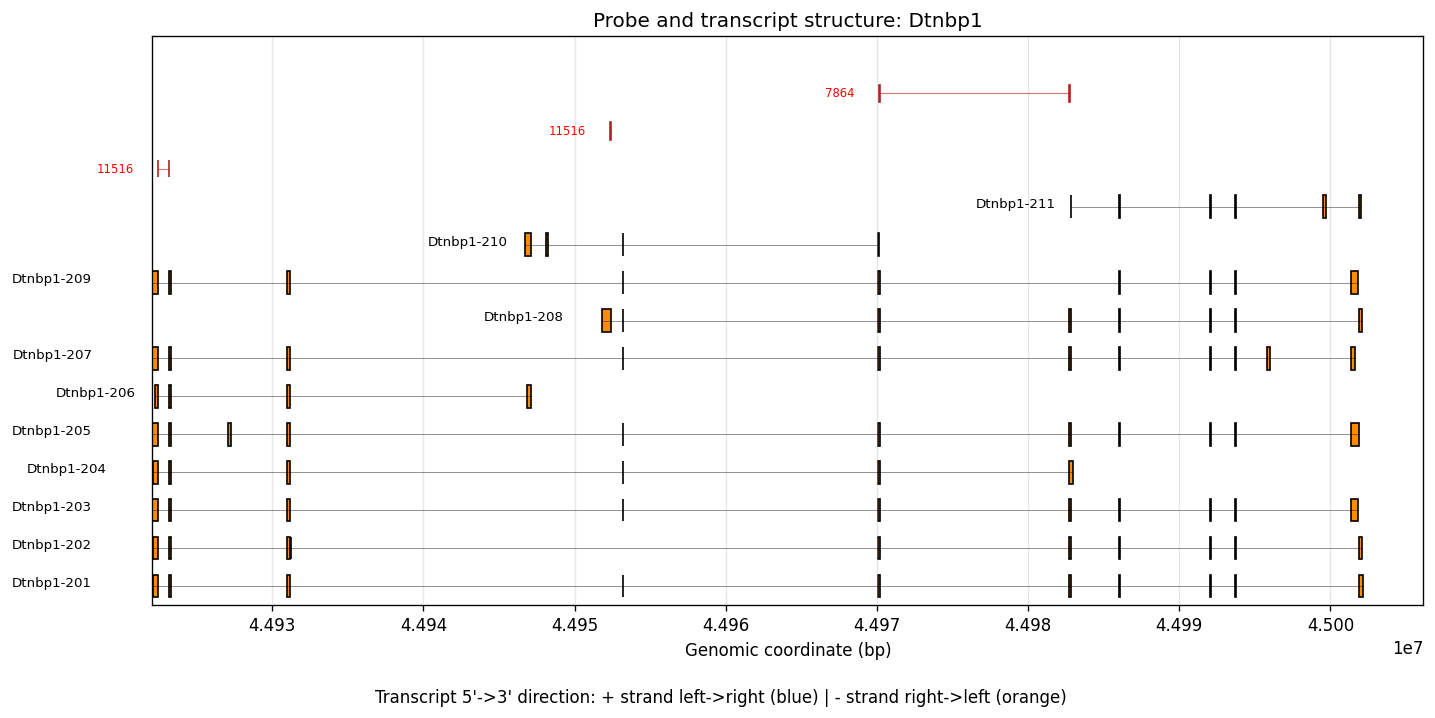
CPU times: user 10.4 s, sys: 55.7 ms, total: 10.4 s
Wall time: 10.5 s
Method comparison: SplisosmFFT vs SplisosmNP#
For a direct method comparison, we run SplisosmNP on the same AnnData table used by SplisosmFFT.
%%time
model_np = SplisosmNP()
model_np.setup_data(
adata=sdata.tables[test_table],
spatial_key='spatial',
layer='counts',
approx_rank=20,
group_iso_by=group_iso_by,
gene_names=gene_name_col,
min_counts=min_counts,
min_bin_pct=min_bin_pct,
)
print(model_np)
=== Non-parametric SPLISOSM model for spatial isoform testings
- Number of genes: 4972
- Number of spots: 144372
- Number of covariates: 0
- Average number of isoforms per gene: 2.091713596138375
=== Test results
- Spatial variability test: NA
- Differential usage test: NA
CPU times: user 8min 6s, sys: 7.83 s, total: 8min 14s
Wall time: 6min 37s
%%time
model_np.test_spatial_variability(
method='hsic-ir',
ratio_transformation='none',
print_progress=True,
)
100%|██████████| 4972/4972 [00:45<00:00, 108.19it/s]
CPU times: user 41.7 s, sys: 16.3 s, total: 58 s
Wall time: 46 s
sv_np = model_np.get_formatted_test_results('sv')[['gene', 'pvalue']].copy()
sv_np = sv_np.rename(columns={'pvalue': 'pvalue_np'})
comparison = sv_res_fft[['gene', 'pvalue']].copy()
comparison = comparison.rename(columns={'pvalue': 'pvalue_fft'})
comparison = comparison.merge(sv_np, on='gene', how='inner')
corr, _ = spearmanr(comparison['pvalue_fft'], comparison['pvalue_np'])
print(f'Genes tested in both methods: {len(comparison)}')
print(f'P-value correlation (Spearman rho): {corr:.4f}')
Genes tested in both methods: 4972
P-value correlation (Spearman rho): 0.7794
fig, ax = plt.subplots(figsize=(5, 5))
x = comparison['pvalue_fft'].to_numpy()
y = comparison['pvalue_np'].to_numpy()
ax.scatter(x + 1e-150, y + 1e-150, s=8, alpha=0.5)
ax.set_xscale('log')
ax.set_yscale('log')
ax.set_xlabel('P-value (SplisosmFFT)')
ax.set_ylabel('P-value (SplisosmNP)')
ax.set_title(f'FFT vs NP ({test_table}, Spearman rho={corr:.2f})')
ax.grid(True, alpha=0.3)
lims = [1e-150, 1.0]
ax.plot(lims, lims, 'r--', alpha=0.5, linewidth=1.5)
ax.set_xlim(lims)
ax.set_ylim(lims)
plt.tight_layout()
plt.show()
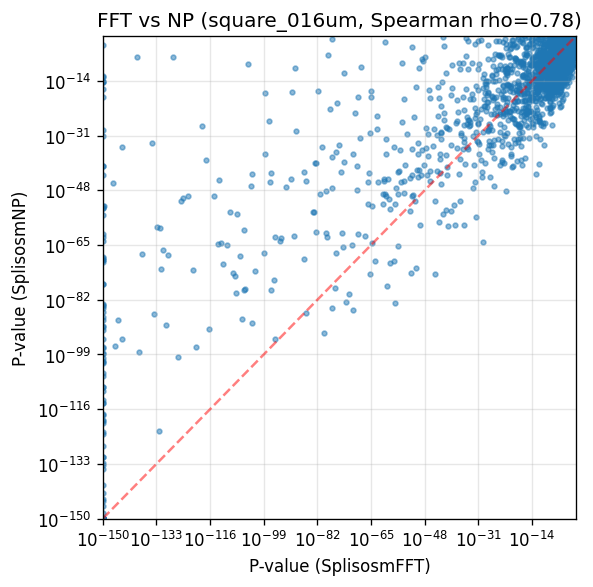
Summary#
load_xenium_codewordenables direct codeword-level multi-resolution binning from Xenium outputs.SplisosmFFTis an efficient default on regular square grids and yield highly similar results toSplisosmNP.
For reproducibility#
import sys
from datetime import date
import splisosm
print('Last updated:', date.today())
print('Python:', sys.version.split()[0])
print('splisosm:', getattr(splisosm, '__version__', 'unknown'))
Last updated: 2026-03-17
Python: 3.12.12
splisosm: 1.0.4